periodiska förlamningar
en kliniskt användbar klassificering av primära periodiska förlamningar, som visas i Tabell 1, inkluderar hypokalemiska, hyperkalemiska och paramyotoniska former.
Tabell 1. Primär periodisk förlamning (modifierad från Jurkat-Rott och Lehmann-Horn ) (öppna tabellen i ett nytt fönster)
|
sjukdom |
Gene |
Protein |
arv |
Mutation |
|
HyperPP |
SCN4A |
Nav1.4 |
Dominant Kvinna |
Fine |
|
NormoPP |
Fine (sabbi-por) |
|||
|
Paramyotoniacongenita |
Fine |
|||
|
HypoPP typ II |
Fine (sabbi-por) |
|||
|
HypoPP för att |
CACNA1S |
Cav1.1 |
Dominant Kvinna |
Gain (sabbi-por) |
|
ThyrotoxicPP |
KCNJ18 |
Kir2.18 |
Dominant Kvinna |
förlust |
|
Andersen-Tawil syndrom |
KCNJ2 |
Kir2.1 |
Dominant Kvinna |
förlust |
den fysiologiska grunden för slapp svaghet är oförmåga hos muskelmembranet (dvs sarcolemma). Förändring av serumkaliumnivån är inte den huvudsakliga defekten i primär PP; den förändrade kaliummetabolismen är ett resultat av PP. I primär och tyrotoxisk PP uppträder slap förlamning med relativt små förändringar i serumkaliumnivån, medan i sekundär PP är serumkaliumnivåerna markant onormala.
ingen enskild mekanism är ansvarig för denna grupp av störningar. Således är de heterogena men delar några vanliga egenskaper. Svagheten är vanligtvis generaliserad men kan lokaliseras. Kranial muskulatur och andningsmuskler sparas vanligtvis. Sträckreflexer är antingen frånvarande eller minskade under attackerna. Muskelfibrerna är elektriskt oföränderliga under attackerna. Muskelstyrkan är normal mellan attacker men efter några år utvecklas en viss grad av fast svaghet i vissa typer av PP (särskilt primär PP). Alla former av primär PP (utom Becker myotonia congenita) är antingen autosomala dominerande ärvda eller sporadiska (troligen härrör från punktmutationer).
spänningskänsliga jonkanaler reglerar nära generering av åtgärdspotentialer (korta och reversibla förändringar av spänningen hos cellmembran). Dessa är selektivt och variabelt permeabla jonkanaler. Energiberoende jontransportörer upprätthåller koncentrationsgradienter. Under genereringen av åtgärdspotentialer rör sig natriumjoner över membranet genom spänningsstyrda jonkanaler. Det vilande muskelfibermembranet polariseras främst genom kloridens rörelse genom kloridkanaler och repolariseras genom kaliumrörelse. Natrium -, kloridoch kalciumkanalopatier, som en grupp, är associerade med myotoni och PP. De funktionella underenheterna av natrium -, kalcium-och kaliumkanaler är homologa. Natriumkanalopatier förstås bättre än kalcium-eller kloridkanalopatier. Alla former av familjär PP visar den slutliga mekanistiska vägen som involverar avvikande depolarisering, inaktiverande natriumkanaler och muskelfiberinexcitabilitet.
diskussionen i denna artikel behandlar främst natrium -, kalcium-och kaliumkanalopatier samt sekundära former av PP. Kloridkanalopatier är inte associerade med episodisk svaghet och diskuteras mer detaljerat i artiklarna om myotoniska störningar.
sammanfattning av kanaldysfunktion i olika typer av PP
med HyperPP snabb kanalinaktivering är mutationer vanligtvis belägna i de inre delarna av transmembransegment eller i de intracellulära slingorna som påverkar dockningsställena för den snabba inaktiverande partikeln, vilket försämrar snabb kanalinaktivering som leder till ihållande Na+ ström.
med HypoPP-hyperpolariseringsaktiverad katjonläckage som motverkar K + – likriktande ström orsakar mutationer yttersta arginin-eller lysinsubstitution.
med NormoPP-depolariseringsaktiverad katjonläcka finns mutationer på djupare platser av spänningssensor för domän II vid kodon R675.
Jonkanaldysfunktion kompenseras vanligtvis väl med normal excitation, och ytterligare triggers är ofta nödvändiga för att producera muskulär oförmåga på grund av långvarig membrandepolarisering.
glukos-och kaliumintag har motsatta effekter vid dessa störningar. I HyperPP utlöser kaliumintag attacken, medan glukos förbättrar den. Däremot framkallar glukos hypokalemiska attacker och kalium är behandlingen för attacken.
notera bilden nedan.
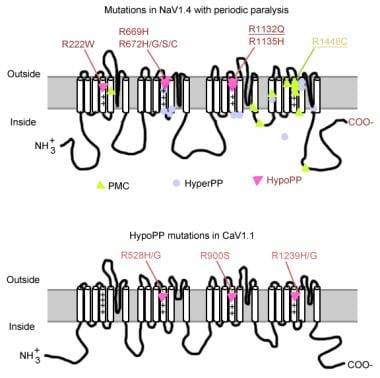 mutationer i periodisk förlamning.
mutationer i periodisk förlamning. Muskelnatriumkanalgen
natriumkanalen har en alfa-subenhet och en beta-subenhet. Alfa-subenheten i natriumkanalen är ett 260-kd glykoprotein innefattande cirka 1800-2000 aminosyror. Denna kanal är mycket bevarad evolutionärt från Drosophila till människa. Den har 4 homologa domäner (I-IV) som viks för att bilda en central pore, var och en med 225-325 aminosyror. Varje domän består av 6 hydrofoba segment (S1-S6) som passerar cellmembranet. Kanalens huvudfunktioner inkluderar spänningskänslig Grind, inaktivering och jonselektivitet. Den extracellulära slingan mellan S5 och S6 doppar in i plasmamembranet och deltar i bildandet av poren. S4-segmentet innehåller positivt laddade aminosyror vid varje tredje position och fungerar som en spänningssensor. Konformationsförändringar kan inträffa under depolarisering, vilket resulterar i aktivering och inaktivering av kanalen. Den cellulära slingan mellan domän III-S6 och domän IV-S1 fungerar som en inaktiverande Grind.
natriumkanalen har 2 grindar (aktivering och inaktivering) och kan existera i 3 tillstånd. I vila med membranet polariserat stängs aktiveringsporten och inaktiveringsporten öppnas. Med depolarisering öppnas aktiveringsporten, vilket tillåter natriumjoner att passera genom jonkanalen och exponerar också en dockningsplats för inaktiveringsporten. Med fortsatt depolarisering stängs inaktiveringsporten, blockerar inträdet av natrium i cellen och får kanalen att komma in i snabbinaktiveringstillståndet. Denna inaktivering av kanalen gör att membranet kan repolariseras, vilket resulterar i en återgång till vilotillstånd med aktiveringsporten stängd och inaktiveringsporten öppnad. Två inaktiveringsprocesser förekommer i skelettmuskulaturen hos däggdjur: snabb inaktivering innebär att åtgärdspotentialen avslutas och verkar på en millisekund tidsskala. Långsam inaktivering tar sekunder till minuter och kan reglera populationen av excitativa natriumkanaler.
natriumkanalmutationer som stör snabb och långsam inaktivering är vanligtvis associerade med en fenotyp av HyperPP och myotoni, där som mutationer som förbättrar långsam eller snabb inaktivering som producerar förlust av natriumkanalfunktion orsakar HypoPP.
mutationer av natriumkanalgenen (SCN4A) har flera allmänna egenskaper. De flesta av mutationerna finns i den ”inaktiverande” länken mellan upprepningar III och IV, i segmentet ”spänningsavkänning” S4 av upprepning IV eller vid det inre membranet där de kan försämra dockningsplatsen för inaktiveringsporten. Den kliniska fenotypen skiljer sig från specifik aminosyrasubstitution och medan viss överlappning kan uppstå mellan Hyperkalemisk PP, paramyotonia congenita (PC) och kaliumförvärrade myotonier (PAM), är de 3 fenotyperna i allmänhet distinkta (som beskrivs nedan). Nästan alla mutanta kanaler har nedsatt snabbinaktivering av natriumström. De flesta patienter är känsliga för systemisk kalium eller för kall temperatur.
två populationer av kanaler finns, mutant och vildtyp; den försämrade snabbinaktiveringen resulterar i långvarig depolarisering av de mutanta muskelfibermembranen och kan förklara de 2 kardinala symtomen på dessa störningar, myotoni och svaghet. I Hyperkalemisk PP uppträder en förstärkning av funktion vid mutantkanalgrindning, vilket resulterar i en ökad natriumström som alltför depolariserar den drabbade muskeln. Mild depolarisering (5-10 mV) av myofibermembranet, som kan orsakas av ökade extracellulära kaliumkoncentrationer, resulterar i att mutantkanalerna bibehålls i det icke-aktiverade läget. Den ihållande inre natriumströmmen orsakar repetitiv avfyrning av natriumkanalerna av vildtyp, vilket uppfattas som styvhet (dvs. myotoni).
om en mer allvarlig depolarisering (20-30 mV) är närvarande, fixeras både normala och onormala kanaler i inaktiveringstillstånd, vilket orsakar svaghet eller förlamning. Således kan subtila skillnader i svårighetsgrad av membrandepolarisering göra skillnaden mellan myotoni och förlamning. Temperaturkänslighet är ett kännetecken för PC. Kall förvärrar myotoni och inducerar svaghet. Ett antal mutationer är associerade med detta tillstånd, 3 av dem på samma plats (1448) i S4-segmentet. Dessa mutationer ersätter arginin med andra aminosyror och neutraliserar denna mycket konserverade S4-positiva laddning. Mutationer av dessa rester är den vanligaste orsaken till PC. Några av de möjliga mekanismerna som är ansvariga för temperaturkänslighet inkluderar följande:
-
temperaturen kan differentiellt påverka konformationsförändringen i mutantkanalen.
-
lägre temperaturer kan stabilisera mutantkanalerna i ett onormalt tillstånd.
-
mutationer kan förändra kanalens känslighet för andra cellulära processer, såsom fosforylering eller andra budbärare.
de flesta fall av Hyperkalemisk PP beror på 2 mutationer i SCN4A, T704M och M1592V. Mutationer i natriumkanalen, särskilt vid rester 1448 och 1313, är ansvariga för paramyotonia congenita. En liten del av hypokalemiska periodiska förlamningsfall är associerade med mutationer vid kodoner 669 och 672 (HypoPP2). I HypoPP2 förbättrar natriumkanalmutationer inaktiveringen för att ge en nettoförlust av funktionsfel.
Normokalemisk PP liknar både HyperPP (kaliumkänslighet) och HypoPP (varaktighet av attacker) och orsakas av SCN4A-mutationer vid en djupare plats för spänningssensor DII vid kodon 675. R675-mutationer skiljer sig från HypoPP genom att dessa mutationer resulterar i depolarisationsaktiverad grindpore som alstrar Bisexuell ström med omvänd spänningsberoende eftersom denna plats utsätts för extracellulära platser vid starkare depolarisering.
Kalciumkanalgen
kalciumkanalgenen (CACNL1A3) är ett komplex av 5 subenheter (alfa-1, alfa-2, beta, gamma och delta). Skelettmuskeldihydropyridinreceptorn (DHP) ligger främst i det tvärgående rörformiga membranet. Alfa – 1-subenheten har bindningsställen för DHP-läkemedel och leder den långsamma kalciumströmmen av L-typ. Det deltar också i excitation-contraction (EC) – koppling och fungerar som en spänningssensor genom dess koppling till ryanodinreceptorn av sarkoplasmatisk retikulum (dvs kalciumfrisättningskanal). Eventuella förändringar i membranpotentialen är kopplade till intracellulär kalciumfrisättning, vilket möjliggör EC-koppling. Punktmutationer i DHP-receptor / kalciumkanal alfa-1-subenhet orsakar hypokalemisk PP (HypoPP1). Två mutationer av cacna1s-genen, R528H och R1239H, är ansvariga för de flesta fall av hypokalemisk PP.
den fysiologiska grunden för sjukdom är fortfarande inte förstådd, men är mer sannolikt på grund av ett misslyckande av excitation snarare än ett misslyckande av EC-koppling. Hypokalemiinducerad depolarisering kan emellertid minska kalciumfrisättningen, vilket påverkar kanalens spänningsreglering direkt eller indirekt genom inaktivering av natriumkanalen. Insulin och adrenalin kan verka på ett liknande sätt. Mutationer av kalciumkanalgenen har vissa likheter med SCN4A-mutationer. Mutationer modifierar kanalinaktivering men inte spänningsberoende aktivering. Inspelningar från myotube-kulturer från drabbade patienter avslöjade en 30% minskning av DHP-känslig l-typ kalciumström. Kanaler inaktiveras vid låga membranpotentialer.
kalciumkanalmutationer orsakar en funktionsförlust som manifesteras som en minskad strömtäthet och långsammare inaktivering. Hur denna inaktivering är relaterad till hypokalemi-inducerade attacker förstås inte. Åtminstone i r528h-mutation uppträder en möjlig sekundär kanalopati, bunden till en minskning av den ATP-känsliga kaliumströmmen från förändrad kalciumhomeostas. De lägre strömmarna associerade med cacnl1a3-mutationer kan något förändra intracellulär kalciumhomeostas, vilket kan påverka egenskaperna och uttrycket av K+ – kanaler, särskilt KATP (ATP-känslig kaliumkanal) som tillhör inåt likriktarklass av kanaler. Insulin verkar också i HypoPP genom att minska denna inre likriktare K+ ström.
Spänningsgivare laddningsförlust står för de flesta fall av HypoPP. Natrium-och kalciumkanaler har homologa porbildande alfa-subenheter. Punktmutationer i CACNL1A3 och SCN4A påverkar argentinska rester i S4-spänningssensorerna i dessa kanaler. Argininmutationer i S4-segment är ansvariga för 90% av HypoPP-Fallen.
Spänningsgivare laddningsförlust står för de flesta fall av HypoPP. Natrium – och kalciumkanaler har homologa porbildande subenheter i CI. Nästan alla mutationer i Cav1.1 (HypoPP-1) och Nav1.4 (HypoPP-2) neutraliserar en positivt laddad aminosyra i en av de yttersta argininerna eller lysinerna hos spänningssensorer. Nav1.4 mutationer är oftast belägna i spänningssensorerna I I, II och III-upprepningar, vilket orsakar en katjonläcka.
Substitution av yttersta arginin med en mindre aminosyra, såsom glycin, öppnar en ledande väg vid hyperpolariserad potential, vilket resulterar i en inre katjonström (katjonläckage eller Kambodjansk ström för att skilja sig från (Kambodjansk-) genom jonledande pore, är en hyperpolariseringsaktiverad ström av monovalenta katjoner genom S4–grindpor som motverkar likriktande k+-strömmar) depolariserande eller destabiliserande vilopotentialen.
S4-segmentet rör sig utåt under depolarisering som stänger den ledande vägen. Muskelfibrer med svåra spänningssensormutationer depolariseras inte bara under hypokalemi utan också vid kaliumnivåer i det normala intervallet, vilket förklarar Interiktal och permanent svaghet. Svår myopati med fettbyte av muskelvävnad finns ofta hos patienter med Cav1.1 R1239H (DIV-mutationer).
glukokortikosteroider orsakar HypoPP genom att stimulera Na + K + ATPas medierat av insulin och amylin.
Kaliumkanalgen
inåtriktad korrigering är en viktig egenskap hos Kir-kanaler. Korrigering innefattar spänningsberoende ledningsporblockering av pore med polyaminer och Mg++ under depolarisering, och denna blockering avlägsnas under potentiell gradient under hyperpolarisering. Kaliumkanalmutationer ses i Andersen-Tawil syndrom och tyrotoxisk PP.
triaden av dysmorfa egenskaper, periodisk förlamning och hjärtarytmier kännetecknar Andersen-Tawil syndrom. Detta syndrom är associerat med mutationer i kcnj2-genen. Kcnj2-genen kodar för den inåtriktade kaliumkanalen Kir2.1. Kaliumkanalmutationer i KCNE3 rapporteras orsaka hypokalemisk PP, men detta har inte underbyggts.
mutationer i Kir2. 6 orsakar mottaglighet för tyrotoxisk PP. Episodisk svaghet som ses i tyrotoxisk PP liknar den som ses i HypoPP och Andersen-Tawil syndrom. Denna sjukdom är vanligast hos asiater och latinamerikanska män. Thyrotoxic PP är en genetisk störning som maskeras av tyrotoxikos. Kir2. 6 uttrycks främst i skelettmuskel. Triiodothyronin förbättrar kcnj18 transkription, vilket kan driva förbättrat uttryck av Kir2.6. PKC aktiveras under tyrotoxikos på grund av ökad pip2-omsättning och Kir-kanaler interagerar direkt med PIP2 under normal grindning. I Andersen-Tawil syndrom är det minskad pip2-affinitet. I tyrotoxisk PP förändrar ingen av mutationerna Kir2.6-rättelse.