Frontiers in Pediatrics
Antecedentes
Un número creciente de estudios han demostrado que RPS6KA3 es una etiología molecular del síndrome de Coffin–Lowry (CLS) (1, 2), un síndrome semidominante ligado al cromosoma X que fue reportado por primera vez por Coffin en 1966 y se caracteriza por baja estatura, dismorfismo facial, discapacidad intelectual grave a profunda (ID), retraso del desarrollo motor, deformidades esqueléticas, trastornos dentales, defectos auditivos y otras deformidades congénitas en hombres, con un intelecto que varía de normal a grave deterioro en hembras heterocigotas (3-6).
La proteína responsable de RPS6KA3 es el polipéptido 3 de la proteína quinasa S6 ribosómica (RSK2), que es una quinasa serina/treonina de una familia de proteínas quinasas activadas por mitógenos (4). La proteína RSK2 se compone de dos dominios quinasas funcionales que se activan de manera secuencial por una serie de fosforilaciones, como el sitio de acoplamiento de PDK (4). Se ha encontrado que la RSK2 está relacionada con la proliferación celular, el bloqueo de la diferenciación celular y la protección de las células de la apoptosis (7, 8). Hasta la fecha, se han notificado casos con síntomas diferentes y con más de 128 mutaciones distintas en el gen RSK2 dividido en 22 exones de chrXp22.2 (RPS6KA3) (4, 5). Recientemente, se ha encontrado que las mutaciones de RPS6KA3 están relacionadas con enfermedades cardíacas, osteosarcoma, compresión de foramen magnum, episodios de caída, etc. (7, 9-12).
En todo el mundo,la prevalencia especulada de CLS puede ser de 1/50, 000-1/100,000 y alrededor del 70-80% de los probandos no tienen antecedentes familiares, mientras que el 20-30% tiene más de un miembro de la familia afectado, según se informó (13). Hasta el momento, no hay cura para esta patología. Aquí, reportamos una mutación de RPS6KA3 en Xp22 en un niño chino. El aspecto fenotípico fue típico, incluyendo retraso en el crecimiento y el desarrollo, como se describió en los informes anteriores.
Presentación del caso
El proband es un niño de 12 meses con dismorfismo facial típico, defecto auditivo y anormalidad ósea (Tabla 1). Nació después de un embarazo normal y dio a luz con un peso al nacer de 2,9 kg (percentil 10) y una longitud de nacimiento de 45 cm (percentil 3) a las 38 semanas, en comparación con los Estándares de Crecimiento Infantil de la OMS en 2006. El aspecto facial presenta frente abultada, orejas prominentes, ojos ampliamente espaciados, fisuras palpebrales inclinadas hacia abajo, nariz corta con columela ancha, alae nasi y tabique gruesos,labio inferior grueso y evertido (Figuras 1A, B). Los dientes deciduos fueron erupcionados a los 8 meses de edad (sin retraso) (Figura 1C). Las manos son cortas, carnosas y con dedos notablemente hiperextensibles que se estrechan de ancho a estrecho con falanges terminales pequeñas y uñas (Figuras 1D, 2D). Pero no había deformidad de su foramen magnum o columna vertebral (Figuras 2B,C). El peso a los 12 meses es de 8,2 kg y la altura es de 68.2 cm (puntuación<-3,17 z, OMS). El metabolismo óseo y el IGF-1α son alteraciones (Vit D 45,2 nmol/L, IGF-1α < 25 ng/ml). Comenzó a sentarse solo a los 9 meses y no podía estar de pie sin ayuda hasta los 12 meses de edad. A los 12 meses de edad, su cociente de inteligencia (CI) era de 56 según los Horarios de Desarrollo de Gesell. Tenía dificultades para permanecer sentado o concentrarse durante la finalización de la tarea. Su umbral auditivo de respuesta auditiva del tronco encefálico (ABR) es >85 db y se diagnostica como un trastorno auditivo. La resonancia magnética (RM) mostró la dilatación de los ventrículos bilaterales y menos materia blanca cerebral (Figura 2A).

Tabla 1. El fenotipo típico del síndrome de Coffin-Lowry de probanda.
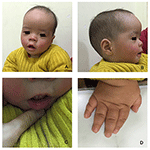
Figura 1. Los dismorfismos faciales distintivos de un chico con síndrome de Coffin-Lowry. Vista lateral de la cara del paciente (B), orejas prominentes, ojos ampliamente espaciados, fisuras palpebrales inclinadas hacia abajo, nariz corta con columela ancha, alae nassi y tabique gruesos,labio inferior grueso y evertado (A, C). (D) Las manos son cortas y con un dedo hinchado y cónico, falanges terminales pequeñas y uñas.
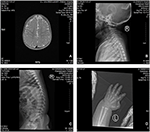
Figura 2. Imágenes por resonancia magnética (IRM) e imágenes de rayos X del caso del síndrome de Coffin-Lowry. A) La resonancia magnética del desarrollo cerebral; B,C) Fotografías de rayos X del canal cervical y espinal. D) Hiperplasia nodular del extremo de la cola del dedo.
Para el análisis genético, se obtuvieron muestras de sangre de la persona. La madre había dado su consentimiento informado a sus hijos. Esta investigación fue aprobada por el comité de bioética para el análisis de genes humanos de la Universidad de Zhejiang.
El análisis de Secuenciación de próxima generación (NGS) se realizó utilizando Agilent Human Genome panel (Agilent Technologies, Inc, Santa Clara, CA, EE.UU.). Se detectó un c. 2185 C > T en chrX20173554 en el probanda, que puede causar p.Mutación Arg729Trp e inestabilidad de RSK2 (Figura 3). El análisis posterior no detectó la misma mutación puntual en otros miembros de la familia, incluidos la madre y el hermano (Material suplementario). Esto indica que la micromutación es nueva pero no heredada de la madre.
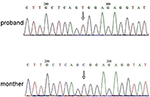
Figura 3. Validación de secuenciación de Sanger para la mutación de RPS6KA3 en el proband en el cromosoma Xp22. Se detectó una mutación de c. 2185 C > T en la región de control de impresión del alelo 290-300, que no se detectó en la madre del proband.
Discusión
El caso que presentamos para la evaluación genética es un niño de 12 meses que nació después de un embarazo sin incidentes de padres sanos. Sin embargo, su edad de desarrollo se retrasó. Al igual que otros pacientes con SCLC afectados, tenía el fenotipo típico observado en el SCLC, que incluía discapacidad intelectual, retraso en el desarrollo motor, estatura pequeña, dedos cónicos, defectos auditivos y rasgos faciales característicos.
RPS6KA3 (OMIM 303600) se sabe que está mutado en pacientes con síndrome de Coffin-Lowry. Exoma c.La variante 2185 C > T en Xp22 era novedosa, separada de la enfermedad, y se predijo que sería perjudicial utilizando cinco programas informáticos in silico diferentes (SIFT, Polyphen, MutationTaster, FATHMM, LRT).
La altura heredada de nuestro proband es de 167,5 cm debido a que la altura de su padre es de 173 cm, mientras que la altura de su madre es de 150 cm (14). Todavía no está claro si la baja estatura del síndrome de Coffin–Lowry podría tratarse con análogos de la hormona del crecimiento. Las RSK son serina / treonina quinasas activadas por ERK / MAPK y constituyen cuatro isoformas (RSK1-4) (15). Los RSK actúan como efectores descendentes de señalización RTK / Ras / ERK (Figura 4) (15-17). Como el gen responsable del CLS, el RSK2 juega un papel importante en la proliferación y migración celular. La activación completa de RSK2 requiere fosforilación en múltiples sitios. Los factores de crecimiento pueden estimular la vía de la señal ERK a RSK2 fosforilado. Como informó Ramos, la activación de ERK / RSK2 está involucrada en la regulación de la migración de células cancerosas (18).
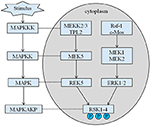
Figura 4. La ruta propuesta de genes relacionados con RSK. Múltiples estímulos patológicos, como la desnutrición intrauterina, desencadenan cascadas de señalización aguas abajo de MAPK, que modulan la proliferación celular y la mineralización ósea.
Además, RSK2 fuera de función está relacionado con la mineralización ósea anormal y también induce malformaciones de columna vertebral y calcificaciones de ligamentum flavum, como lordosis torácica, escoliosis, cifosis y enfermedad degenerativa del disco (19, 20). Informes anteriores identificaron el ERK / RSK2 como regulador de la formación ósea in vivo (7). De hecho, como Marques et al. los ratones con deficiencia de RSK2 recapitularon la pérdida ósea progresiva debido a la disminución de la mineralización ósea por parte de los osteoblastos observada en pacientes con SLC (21). Aunque la diferenciación de estas células in vitro se bloqueó drásticamente, el fenotipo in vivo se atribuyó claramente a una disminución celular autónoma de la actividad de los osteoblastos más que a una disminución de su número (7). Curiosamente, recientemente se demostró que RSK2 interactúa con TNF-RI en la diferenciación de osteoblastos afectados (18, 22). Como sabemos, la hormona del crecimiento puede causar un aumento de la densidad mineral ósea. El progreso puede agravar las calcificaciones del ligamento flavum y la deformidad esquelética (23).
Observaciones finales
El presente estudio es el primero en reportar un caso chino de SCL con mutación de RPS6KA3, así como retraso en el crecimiento, anomalías faciales múltiples, discapacidades intelectuales y motoras. Se especula que la aplicación de análogos de la hormona del crecimiento puede aumentar la incidencia y la invasividad de los cánceres, así como el riesgo de malformación de la columna vertebral. Si no se confirma su eficacia y seguridad, se debe considerar con especial cautela la aplicación de análogos de la hormona del crecimiento en pacientes con SCL. Los estudios a largo plazo con resultados relevantes serán esenciales en la investigación clínica futura.
Consentimiento del paciente
Obtuvimos el consentimiento informado para publicar este reporte de caso y para usar imágenes relacionadas de los padres del paciente.
Contribuciones de los autores
JS contribuyó sustancialmente a la concepción de este manuscrito. YL, LZ, JZ y DW contribuyeron a la adquisición, análisis o interpretación de datos. YL redactó el manuscrito. JS revisó críticamente el manuscrito en busca de contenido intelectual importante.
Financiación
Este trabajo fue apoyado por subvenciones de la Fundación Nacional de Ciencias Naturales de China (no. 81501293 y 81773440). Reconocemos a la familia del caso descrito en este informe que conoce y aprueba esta cuenta no identificada.
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un conflicto de intereses potencial.
El revisor BP y el Editor responsable declararon su afiliación compartida en el momento de la revisión.
Material suplementario
Figura S1. Validación de secuenciación de Sanger para la mutación de RPS6KA3 en el hermano del proband.
1. Stornetta RL, Zhu JJ. Señalización Ras y Rap en plasticidad sináptica y trastornos mentales. Neuroscientist (2011) 17:54-78. doi: 10.1177/1073858410365562
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
6. Knish LV. Efecto de las preparaciones de vitamina E utilizadas en el embarazo sobre la placenta. Pediatr Akus Ginekol. (1966) 2:51–5.
Resumen de PubMed | Google Scholar
10. Gschwind M, Foletti G, Baumer A, Bottani A, Novy J. Estado epiléptico no convulsivo recurrente en un paciente con síndrome de coffin-lowry. Síndrome de Mol. (2015) 6:91–5. doi: 10.1159/000430429
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
12. Martinez HR, Niu MC, Sutton VR, Pignatelli R, Vatta M, Jefferies JL. Síndrome de Ataúd-Lowry y miocardiopatía no compactada ventricular izquierda con un patrón restrictivo. Am J Med Genet A (2011) 155A:3030–4. doi: 10.1002 / ajmg.a. 33856
Resumen de PubMed | Texto Completo de CrossRef / Google Scholar
14. Cameron N. Programas básicos para la evaluación de la madurez esquelética y la predicción de la estatura adulta utilizando el método Tanner-Whitehouse. Ann Hum Biol. (1984) 11:261–4. doi: 10.1080/03014468400007151
Resumen de PubMed / Texto Completo de CrossRef | Google Scholar
15. Romeo Y, Zhang X, Roux PP. Regulación y función de la familia de proteínas quinasas RSK. Biochem J. (2012) 441:553-69. doi: 10.1042 | BJ20110289
Resumen de PubMed / Texto completo de CrossRef / Google Scholar
17. Beck K, Ehmann N, Andlauer TF, Ljaschenko D, Strecker K, Fischer M, et al. La pérdida del gen RSK2 asociado al síndrome de Coffin-Lowry altera la actividad ERK, la función sináptica y el transporte axonal en motoneuronas de Drosophila. Dis Model Mech. (2015) 8:1389–400. doi: 10.1242 / dmm.021246
Resumen de PubMed / Texto Completo de CrossRef / Google Scholar
18. Shi GX, Yang WS, Jin L, Matter ML, Ramos JW. RSK2 impulsa la motilidad celular mediante la fosforilación de serina de GRANDES y la activación de GTPasas Rho. Proc Natl Acad Sci USA. (2018) 115:E190–9. doi: 10.1073 / pnas.1708584115
Resumen de PubMed / Texto completo Cruzado / Google Scholar