frontiere în pediatrie
context
un număr tot mai mare de studii au demonstrat că RPS6KA3 este o etiologie moleculară a sindromului Coffin–Lowry (CLS) (1, 2), un sindrom semidominant legat de X care a fost raportat pentru prima dată de Coffin în 1966 și caracterizat prin statură scurtă, dismorfism facial, dizabilitate intelectuală severă până la profundă (ID), întârziere deformări scheletice, tulburări dentare, defecte de auz și alte deformări congenitale la bărbați, cu intelect variază de la normal la sever afectarea la femelele heterozigote (3-6).
proteina responsabilă de RPS6KA3 este proteina ribozomală S6 kinază polipeptidă 3 (RSK2), care este o serină/treonin kinază a unei familii de proteine kinaze activate de mitogen (4). Proteina RSK2 este compusă din două domenii funcționale de kinază care sunt activate în mod secvențial printr-o serie de fosforilări, cum ar fi site-ul de andocare PDK (4). S-a constatat că RSK2 este legat de stimularea proliferării celulare, blocarea diferențierii celulare și protejarea celulelor de apoptoză (7, 8). Până în prezent, au fost raportate cazuri cu simptome diferite și cu peste 128 de mutații distincte ale genei RSK2 împărțite în 22 de exoni ai chrXp22.2 (RPS6KA3) (4, 5). Recent, mutațiile RPS6KA3 s-au dovedit a fi legate de boli de inimă, osteosarcom, compresie foramen magnum, episoade de picătură și așa mai departe (7, 9-12).
la nivel mondial, prevalența speculată a CLS poate fi de 1/50.000-1 / 100.000 și aproximativ 70-80% dintre probandi nu au antecedente familiale,în timp ce 20-30% au mai mult de un membru al familiei afectat, după cum s-a raportat (13). Până în prezent, nu există nici un remediu pentru această patologie. Aici, raportăm o mutație a RPS6KA3 la Xp22 la un băiat chinez. Aspectul fenotipic a fost tipic, inclusiv întârzierea creșterii și dezvoltării, așa cum a fost descris în rapoartele anterioare.
prezentarea cazului
probandul este un băiat de 12 luni cu dismorfism facial tipic, defect auditiv și anomalie osoasă (Tabelul 1). S-a născut după o sarcină normală și a născut cu o greutate la naștere de 2,9 kg (percentila a 10-a) și o lungime la naștere de 45 cm (percentila a 3-A) la 38 de săptămâni, comparativ cu standardele OMS de creștere a copilului în 2006. Aspectul facial prezintă frunte bombată, urechi proeminente, ochi larg distanțați, fisuri palpebrale înclinate în jos, nas scurt cu columelă largă, alae nasi gros și sept,alunecare groasă și evertată (figurile 1A, B). Dinții de foioase au fost erupți la vârsta de 8 luni (nu întârziați) (figura 1C). Mâinile sunt scurte, cărnoase și cu degete remarcabil de hiperextensibile, care se îngustează de la larg la îngust cu mici falange terminale și unghii (figurile 1D, 2D). Dar nu a existat nici o deformare a foramen magnum sau a coloanei vertebrale (figurile 2B, C). Greutatea la 12 luni este de 8,2 kg, iar înălțimea este de 68.2 cm (<-3.17 Z scor, OMS). Metabolizarea osoasă și IGF-1 inqc este perturbată (Vit D 45,2 nmol/L, IGF-1 inqc < 25 ng/mL). A început să stea singur la 9 luni și nu a putut sta fără ajutor până la vârsta de 12 luni. La vârsta de 12 luni, coeficientul său de inteligență (IQ) era de 56, conform programelor de dezvoltare Gesell. A avut dificultăți să rămână așezat sau să se concentreze în timpul finalizării sarcinii. Pragul său auditiv de răspuns auditiv al trunchiului cerebral (ABR) este >85 db și este diagnosticat ca o tulburare de auz. Imagistica prin rezonanță magnetică (RMN) a arătat dilatarea ventriculilor bilaterali și mai puțină substanță albă cerebrală (figura 2a).

Tabelul 1. Fenotipul tipic al sindromului Coffin-Lowry al probandului.
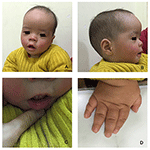
Figura 1. Dismorfismele faciale distinctive ale unui băiat cu sindromul Coffin–Lowry. O vedere laterală a feței pacientului( B), urechi proeminente, ochi larg distanțați, fisuri palpebrale înclinate în jos, nas scurt cu columelă largă, alae nasi gros și sept,subțire groasă și evertată (A, C). (D) mâinile sunt scurte și cu degetul conic pufos, falangele terminale mici și unghiile.
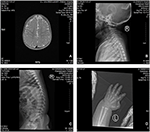
Figura 2. Imagistica prin rezonanță magnetică (RMN) și imaginile cu raze X ale cazului sindromului Coffin-Lowry. (A) RMN-ul dezvoltării cerebrale; (B,C) fotografii cu raze X ale canalului Cervical și spinal. (D) hiperplazia nodulară a capătului cozii degetului.
pentru analiza genetică, probele de sânge au fost obținute de la individ. Mama și-a dat consimțământul în cunoștință de cauză pentru copiii ei. Această cercetare a fost aprobată de Comitetul de Bioetică pentru analiza genelor umane de la Universitatea Zhejiang.
analiza secvențierii de generație următoare (NGS) a fost efectuată utilizând panoul genomului uman Agilent (Agilent Technologies, Inc, Santa Clara, CA, SUA). Un c.2185 c > T la chrX20173554 a fost detectat în proband, ceea ce poate provoca p.Mutația Arg729Trp și instabilitatea RSK2 (Figura 3). Analiza ulterioară nu a detectat aceeași mutație punctuală la alți membri ai familiei, inclusiv mama, fratele (Material suplimentar). Acest lucru a indicat că micromutarea este nouă, dar nu moștenită de la mamă.
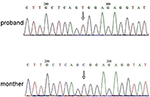
Figura 3. Validarea secvențierii Sanger pentru mutația RPS6KA3 în probandul la cromozomul Xp22. O mutație a c. 2185 c > T a fost detectată în regiunea de control a imprimării din alela 290-300, care nu a fost detectată la mama lui proband.
discuție
cazul pe care îl prezentăm pentru evaluarea geneticii este un băiat de 12 luni care s-a născut după o sarcină fără evenimente de la părinți sănătoși. Cu toate acestea, vârsta sa de dezvoltare a fost întârziată. Ca și alți pacienți cu CLS afectați, el a avut fenotipul tipic observat în CLS, inclusiv dizabilitate intelectuală, dezvoltare motorie întârziată, statură mică, degete conice, defecte de auz și trăsături faciale caracteristice.
RPS6KA3 (OMIM 303600) este cunoscut ca fiind mutant la pacienții cu sindrom Coffin-Lowry. Exom c.Varianta 2185 c > t la Xp22 a fost nouă, separată de boală și s-a prezis că va fi dăunătoare folosind cinci programe diferite in silico (SIFT, Polyphen, MutationTaster, FATHMM, LRT).
înălțimea moștenită a probandului nostru este de 167,5 cm, deoarece înălțimea tatălui său este de 173 cm, în timp ce înălțimea mamei sale este de 150 cm (14). Nu este încă clar dacă statura scurtă a sindromului Coffin–Lowry ar putea fi tratată cu analogi ai hormonului de creștere. RSK–urile sunt serină/treonin kinaze activate de ERK/MAPK și constituie patru izoforme (RSK1-4) (15). RSK-urile acționează ca efectori în aval ai semnalizării RTK/Ras/ERK (Figura 4) (15-17). Ca gena responsabilă pentru CLS, RSK2 joacă un rol important în proliferarea și migrarea celulelor. Activarea completă a RSK2 necesită fosforilare la mai multe site-uri. Factorii de creștere pot stimula calea semnalului ERK către RSK fosforilat2. După cum a raportat Ramos, activarea ERK / RSK2 este implicată în reglarea migrării celulelor canceroase (18).
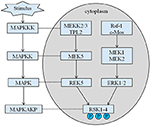
Figura 4. Calea propusă a genelor legate de RSK. Stimuli patologici multipli, cum ar fi malnutriția intrauterină, declanșează cascade de semnalizare în aval ale MAPK, care modulează proliferarea celulară și mineralizarea osoasă.
mai mult, funcția RSK2 este legată de mineralizarea osoasă anormală și, de asemenea, induce malformații ale coloanei vertebrale și calcificări ale ligamentum flavum, cum ar fi lordoza toracică, scolioza, cifoza și boala degenerativă a discului (19, 20). Rapoartele anterioare au identificat ERK / RSK2 ca regulator al formării osoase in vivo (7). Într-adevăr, as Marques și colab. raportat, șoarecii cu deficit de RSK2 au recapitulat pierderea osoasă progresivă datorată scăderii mineralizării osoase de către osteoblastele observate la pacienții cu CLS (21). Deși diferențierea acestor celule in vitro a fost blocată drastic, fenotipul in vivo a fost atribuit în mod clar unei scăderi autonome celulare a activității osteoblastelor, mai degrabă decât unei scăderi a numărului acestora (7). Interesant este că rsk2 a fost recent demonstrat că interacționează cu TNF-RI la diferențierea osteoblastelor afectate (18, 22). După cum știm, hormonul de creștere poate provoca o densitate minerală osoasă crescută. Progresul poate agrava calcificările ligamentum flavum și deformarea scheletului (23).
observații finale
studiul de față este primul care a raportat un caz chinez de CLS cu mutație a RPS6KA3, precum și o întârziere distinctă a creșterii, anomalii faciale multiple, dizabilități intelectuale și motorii. Speculăm că aplicarea analogilor hormonului de creștere poate crește incidența și invazivitatea cancerelor, precum și riscul de malformație a coloanei vertebrale. Dacă eficacitatea și siguranța acestuia nu sunt confirmate, aplicarea analogilor hormonului de creștere la pacienții cu CLS trebuie luată în considerare cu precauție deosebită. Studiile pe termen lung cu rezultate relevante vor fi esențiale în viitoarele cercetări clinice.
consimțământul pacientului
am obținut consimțământul informat pentru publicarea acestui raport de caz și pentru utilizarea imaginilor conexe de la părinții pacientului.
contribuțiile autorului
JS au contribuit substanțial la conceperea acestui manuscris. YL, LZ, JZ și DW au contribuit la achiziționarea, analiza sau interpretarea datelor. YL a redactat manuscrisul. JS a revizuit critic manuscrisul pentru un conținut intelectual important.
finanțare
această lucrare a fost susținută de granturi de la Fundația Națională de științe Naturale din China (nr. 81501293 și 81773440). Recunoaștem familia cazului descris în acest raport care cunoaște și aprobă acest cont de-identificat.
Declarație privind conflictul de interese
autorii declară că cercetarea a fost realizată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
recenzorul BP și editorul de manipulare și-au declarat afilierea comună la momentul revizuirii.
Material Suplimentar
Figura S1. Validarea secvențierii Sanger pentru mutația RPS6KA3 în fratele probandului.
1. Stornetta RL, Zhu JJ. Semnalarea Ras și Rap în plasticitatea sinaptică și tulburările mentale. Neurolog (2011) 17:54-78. doi: 10.1177/1073858410365562
PubMed rezumat / CrossRef Text Complet / Google Scholar
6. Knish LV. efectul preparatelor de vitamina E utilizate în timpul sarcinii asupra placentei. Pediatrul Akus Ginekol. (1966) 2:51–5.
Rezumat PubMed / Google Scholar
10. Gschwind M, Foletti G, Baumer A, Bottani A, Novy J. status epileptic recurent nonconvulsiv la un pacient cu sindrom coffin-lowry. Sindromul Mol. (2015) 6:91–5. doi: 10.1159/000430429
PubMed rezumat / CrossRef Text Complet / Google Scholar
12. Martinez HR, Niu MC, Sutton VR, Pignatelli R, Vatta M, Jefferies JL. Sindromul Coffin-Lowry și cardiomiopatia necompactantă a ventriculului stâng cu un model restrictiv. Am J Med Genet A (2011) 155A:3030–4. doi: 10.1002 / ajmg.a. 33856
PubMed rezumat / CrossRef Text Complet / Google Scholar
14. Cameron N. programe de bază pentru evaluarea maturității scheletice și predicția înălțimii adulților folosind metoda Tanner-Whitehouse. Ann Hum Biol. (1984) 11:261–4. doi: 10.1080/03014468400007151
PubMed rezumat / CrossRef Text Complet / Google Scholar
15. Romeo Y, Zhang X, Roux PP. Reglarea și funcția familiei RSK de protein kinaze. Biochem J. (2012) 441:553-69. doi: 10.1042 / BJ20110289
PubMed rezumat / CrossRef Text Complet / Google Scholar
17. Beck K, Ehmann N, Andlauer TF, Ljaschenko D, Strecker K, Fischer M și colab. Pierderea genei asociate sindromului Coffin-Lowry rsk2 modifică activitatea ERK, funcția sinaptică și transportul axonal în motoneuronii Drosophila. Dis Model Mech. (2015) 8:1389–400. doi: 10.1242 / dmm.021246
PubMed Rezumat / CrossRef Text Complet / Google Scholar
18. Shi GX, Yang WS, Jin L, materie ML, Ramos JW. RSK2 conduce motilitatea celulară prin fosforilarea serină a LARG și activarea Gtpazelor Rho. Proc Natl Acad Sci Statele Unite ale Americii. (2018) 115:E190–9. doi: 10.1073 / pnas.1708584115
PubMed Rezumat / CrossRef Text Complet / Google Scholar