Grenzen in der Pädiatrie
Hintergrund
Eine wachsende Anzahl von Studien hat gezeigt, dass RPS6KA3 eine molekulare Ätiologie des Coffin–Lowry-Syndroms (CLS) ist (1, 2), ein X-chromosomales semidominantes Syndrom, das erstmals 1966 von Coffin berichtet wurde und durch Kleinwuchs, Gesichtsdysmorphismus, schwere bis tiefe geistige Behinderung (ID), motorische Entwicklungsverzögerung , progressive Skelettdeformitäten, Zahnerkrankungen, Hörfehler und andere angeborene Deformitäten bei Männern, mit Intellekt reicht von normal bis schwer beeinträchtigt bei heterozygoten Weibchen (3-6).
Das für RPS6KA3 verantwortliche Protein ist das ribosomale Protein S6-Kinase-Polypeptid 3 (RSK2), das eine Serin/Threonin-Kinase einer Familie von mitogenaktivierten Proteinkinasen ist (4). Das RSK2-Protein besteht aus zwei funktionellen Kinasedomänen, die nacheinander durch eine Reihe von Phosphorylierungen aktiviert werden, wie z. B. die PDK-Andockstelle (4). Es wurde festgestellt, dass RSK2 mit der Aufforderung zur Zellproliferation, der Blockierung der Zelldifferenzierung und dem Schutz der Zellen vor Apoptose zusammenhängt (7, 8). Bisher gab es Berichte über Fälle mit unterschiedlichen Symptomen und mit über 128 verschiedenen Mutationen im RSK2-Gen, aufgeteilt in 22 Exons von chrXp22.2 (RPS6KA3) (4, 5). Kürzlich wurde festgestellt, dass die Mutationen von RPS6KA3 mit Herzerkrankungen, Osteosarkomen, Foramen Magnum-Kompression, Drop-Episoden usw. zusammenhängen (7, 9-12).
Weltweit kann die spekulierte Prävalenz von CLS 1/50.000-1 / 100.000 betragen, und etwa 70-80% der Probanden haben keine Familienanamnese, während 20-30% mehr als ein betroffenes Familienmitglied haben, wie berichtet (13). Bisher gibt es keine Heilung für diese Pathologie. Hier berichten wir über eine Mutation von RPS6KA3 bei Xp22 in einem chinesischen Jungen. Das phänotypische Erscheinungsbild war typisch, einschließlich Wachstums- und Entwicklungsverzögerung, wie in den vorherigen Berichten beschrieben.
Falldarstellung
Der Proband ist ein 12 Monate alter Junge mit typischem Gesichtsdysmorphismus, Hördefekt und Knochenanomalie (Tabelle 1). Er wurde nach einer normalen Schwangerschaft geboren und mit einem Geburtsgewicht von 2,9 kg (10. Perzentil) und einer Geburtslänge von 45 cm (3. Perzentil) nach 38 Wochen im Vergleich zu den WHO-Kinderwachstumsstandards im Jahr 2006 zur Welt gebracht. Das Gesichtsbild zeigt eine gewölbte Stirn, hervorstehende Ohren, weit auseinander liegende Augen, nach unten geneigte Palpebralfissuren, kurze Nase mit breiter Columella, dicke Alae Nasi und Septum, dicke und umgestülpte Unterlippe (Abbildungen 1A, B). Die Milchzähne waren im Alter von 8 Monaten (nicht verzögert) durchgebrochen (Abbildung 1C). Die Hände sind kurz, fleischig und mit bemerkenswert überstreckbaren Fingern, die sich von breit zu schmal verjüngen, mit kleinen Endphalangen und Nägeln (Abbildungen 1D, 2D). Es gab jedoch keine Deformität seines Foramen magnum oder seiner Wirbelsäule (Abbildungen 2B, C). Das Gewicht nach 12 Monaten beträgt 8,2 kg und die Körpergröße beträgt 68.2 cm (<-3,17 z-Score, WER). Der Knochenstoffwechsel und IGF-1α ist Störung (Vit D 45,2 nmol / L, IGF-1α < 25 ng / ml). Er begann mit 9 Monaten alleine zu sitzen und konnte erst im Alter von 12 Monaten ohne Hilfe stehen. Im Alter von 12 Monaten betrug sein Intelligenzquotient (IQ) gemäß den Gesell-Entwicklungsplänen 56. Er hatte Schwierigkeiten, während der Erledigung der Aufgabe sitzen zu bleiben oder sich zu konzentrieren. Seine Hörschwelle der auditorischen Hirnstammreaktion (ABR) beträgt > 85 dB und wird als Hörstörung diagnostiziert. Die Magnetresonanztomographie (MRT) zeigte die Erweiterung der bilateralen Ventrikel und weniger zerebrale weiße Substanz (Abbildung 2A).

Tabelle 1. Der typische Phänotyp des Coffin-Lowry-Syndroms des Probanden.
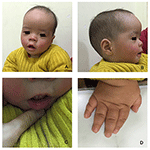
Abbildung 1. Die charakteristischen Gesichtsdysmorphismen eines Jungen mit Coffin-Lowry-Syndrom. Eine Seitenansicht des Gesichts des Patienten (B), abstehende Ohren, weit auseinander liegende Augen, nach unten geneigte Palpebralfissuren, kurze Nase mit breiter Columella, dicke Alae nasi und Septum, dicke und umgestülpte Unterlippe (A, C). (D) Die Hände sind kurz und mit geschwollenen, sich verjüngenden Fingern, kleinen Endphalangen und Nägeln.
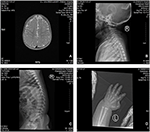
Abbildung 2. Die Magnetresonanztomographie (MRT) und Röntgenbilder des Coffin-Lowry–Syndroms. (A) Die MRT der zerebralen Entwicklung; (B, C) Röntgenaufnahmen des Gebärmutterhalses und des Spinalkanals. (D) Knotige Hyperplasie des Fingerschwanzendes.
Für die genetische Analyse wurden Blutproben von der Person erhalten. Die Mutter hatte Einverständniserklärung für ihre Kinder gegeben. Diese Forschung wurde vom Bioethik-Komitee für menschliche Genanalyse an der Zhejiang-Universität genehmigt.
Die Next Generation Sequencing (NGS)-Analyse wurde mit dem Agilent Human Genome Panel (Agilent Technologies, Inc, Santa Clara, CA, USA) durchgeführt. Ein c.2185 C > T bei chrX20173554 wurde im Probanden nachgewiesen, was p verursachen kann.Arg729Trp-Mutation und RSK2-Instabilität (Abbildung 3). Die anschließende Analyse ergab nicht die gleiche Punktmutation bei anderen Familienmitgliedern, einschließlich Mutter, Bruder (Ergänzendes Material). Dies deutete darauf hin, dass die Mikromutation neu ist, aber nicht von der Mutter geerbt wurde.
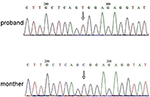
Abbildung 3. Sanger-Sequenzierungsvalidierung für die Mutation von RPS6KA3 im Probanden auf Chromosom Xp22. Eine Mutation von c.2185 C > T wurde in der Prägungskontrollregion von Allel 290-300 nachgewiesen, die bei der Mutter des Probanden nicht nachgewiesen wurde.
Diskussion
Der Fall, den wir für die genetische Bewertung präsentieren, ist ein 12 Monate alter Junge, der nach einer ereignislosen Schwangerschaft von gesunden Eltern geboren wurde. Sein Entwicklungsalter verzögerte sich jedoch. Wie andere betroffene CLS-Patienten hatte er den typischen Phänotyp, der bei CLS beobachtet wurde, einschließlich geistiger Behinderung, verzögerter motorischer Entwicklung, kleiner Statur, sich verjüngender Finger, Hördefekt und charakteristischer Gesichtszüge.
Es ist bekannt, dass RPS6KA3 (OMIM 303600) bei Patienten mit Coffin-Lowry-Syndrom mutiert ist. Exom c.2185 C > T-Variante bei Xp22 war neu, getrennt von der Krankheit, und wurde vorhergesagt, schädlich zu sein, mit fünf verschiedenen in Silico-Software (SIFT, Polyphenol, MutationTaster, FATHMM, LRT).
Die vererbte Größe unseres Probanden beträgt 167,5 cm, da die Größe seines Vaters 173 cm beträgt, während die Größe seiner Mutter 150 cm beträgt (14). Ob die Kleinwüchsigkeit des Coffin-Lowry-Syndroms mit Wachstumshormon-Analoga behandelt werden könnte, ist noch nicht klar. RSKs sind durch ERK/MAPK aktivierte Serin / Threonin-Kinasen und bilden vier Isoformen (RSK1–4) (15). RSKs fungieren als nachgeschaltete Effektoren der RTK / Ras / ERK-Signalisierung (Abbildung 4) (15-17). Als das für CLS verantwortliche Gen spielt RSK2 eine wichtige Rolle bei der Zellproliferation und -migration. Die vollständige Aktivierung von RSK2 erfordert eine Phosphorylierung an mehreren Stellen. Wachstumsfaktoren können den ERK-Signalweg zu phosphoryliertem RSK2 stimulieren. Wie Ramos berichtete, ist die Aktivierung von ERK / RSK2 an der Regulierung der Migration von Krebszellen beteiligt (18).
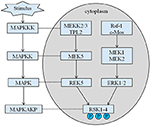
Abbildung 4. Der vorgeschlagene Weg von RSK-verwandten Genen. Mehrere pathologische Reize, wie intrauterine Unterernährung, lösen nachgeschaltete Signalkaskaden von MAPK aus, die die Zellproliferation und Knochenmineralisierung modulieren.
Darüber hinaus hängt RSK2 außer Funktion mit einer abnormalen Knochenmineralisierung zusammen und induziert auch Fehlbildungen der Wirbelsäule und Verkalkungen des Ligamentum flavum wie Thoraxlordose, Skoliose, Kyphose und degenerative Bandscheibenerkrankungen (19, 20). Frühere Berichte identifizierten den ERK / RSK2 als Regulator der Knochenbildung in vivo (7). In der Tat, wie Marques et al. berichtet, rekapitulierten RSK2-defiziente Mäuse den fortschreitenden Knochenverlust aufgrund einer verminderten Knochenmineralisierung durch die Osteoblasten, die bei Patienten mit CLS beobachtet wurden (21). Obwohl die Differenzierung dieser Zellen in vitro drastisch blockiert war, wurde der In-vivo-Phänotyp eindeutig einer zellautonomen Abnahme der Aktivität der Osteoblasten und nicht einer Abnahme ihrer Anzahl zugeschrieben (7). Interessanterweise wurde kürzlich gezeigt, dass RSK2 mit TNF-RI interagiert, um die Differenzierung von Osteoblasten zu beeinflussen (18, 22). Wie wir wissen, kann Wachstumshormon eine erhöhte Knochenmineraldichte verursachen. Der Fortschritt kann Verkalkungen von Ligamentum flavum und Skelettdeformität verschlimmern (23).
Abschließende Bemerkungen
Die vorliegende Studie ist die erste, die einen chinesischen Fall von CLS mit Mutation von RPS6KA3 sowie einer deutlichen Wachstumsverzögerung, multiplen Gesichtsanomalien, intellektuellen und motorischen Behinderungen berichtet. Wir spekulieren, dass die Anwendung von Wachstumshormon-Analoga die Inzidenz und Invasivität von Krebserkrankungen sowie das Risiko von Wirbelsäulenfehlbildungen erhöhen kann. Wenn seine Wirksamkeit und Sicherheit nicht bestätigt werden, sollte die Anwendung von Wachstumshormonanaloga bei Patienten mit CLS besonders vorsichtig in Betracht gezogen werden. Langzeitstudien mit relevanten Ergebnissen werden in der zukünftigen klinischen Forschung unerlässlich sein.
Einwilligung des Patienten
Wir haben die Einwilligung nach Aufklärung zur Veröffentlichung dieses Fallberichts und zur Verwendung verwandter Bilder von den Eltern des Patienten eingeholt.
Autorenbeiträge
JS hat wesentlich zur Konzeption dieses Manuskripts beigetragen. YL, LZ, JZ und DW trugen zur Erfassung, Analyse oder Interpretation von Daten bei. YL entwarf das Manuskript. JS überarbeitete das Manuskript kritisch auf wichtige intellektuelle Inhalte.
Finanzierung
Diese Arbeit wurde durch Zuschüsse der National Natural Science Foundation of China (Nr. 81501293 und 81773440) unterstützt. Wir erkennen die in diesem Bericht beschriebene Familie des Falles an, die dieses de-identifizierte Konto kennt und genehmigt.
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Der Reviewer BP und der Editor erklärten zum Zeitpunkt der Überprüfung ihre gemeinsame Zugehörigkeit.
Ergänzendes Material
Abbildung S1. Sanger-Sequenzierungsvalidierung für die Mutation von RPS6KA3 im Bruder des Probanden.
1. In: Stornetta RL, Zhu JJ. Ras- und Rap-Signalisierung bei synaptischer Plastizität und psychischen Störungen. Neurowissenschaftler (2011) 17: 54-78. doi: 10.1177/1073858410365562
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
6. Knish LV. Wirkung von Vitamin E-Präparaten in der Schwangerschaft auf die Plazenta. Pediatr Akus Ginekol. (1966) 2:51–5.
PubMed Zusammenfassung / Google Scholar
10. Gschwind M, Foletti G, Baumer A, Bottani A, Novy J. Rezidivierender nicht krampfhafter Status epilepticus bei einem Patienten mit Coffin-Lowry-Syndrom. In: Mol Syndromol. (2015) 6:91–5. doi: 10.1159/000430429
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
12. Martinez HR, Niu MC, Sutton VR, Pignatelli R, Vatta M, Jefferies JL. Coffin-Lowry-Syndrom und linksventrikuläre Nichtkompaktions-Kardiomyopathie mit restriktivem Muster. Am J Mit Genet A (2011) 155A:3030–4. doi: 10.1002/ajmg.a.33856
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
14. Cameron N. GRUNDLEGENDE Programme zur Beurteilung der Skelettreife und zur Vorhersage der Körpergröße von Erwachsenen nach der Tanner-Whitehouse-Methode. In: Ann Hum Biol. (1984) 11:261–4. doi: 10.1080/03014468400007151
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
15. Romeo Y, Zhang X, Mehlschwitze PP. Regulation und Funktion der RSK-Familie von Proteinkinasen. Biochem J. (2012) 441: 553-69. doi: 10.1042/BJ20110289
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
17. Beck K, Ehmann N, Andlauer TF, Ljaschenko D, Strecker K, Fischer M, et al. Der Verlust des mit dem Coffin-Lowry-Syndrom assoziierten Gens RSK2 verändert die ERK-Aktivität, die synaptische Funktion und den axonalen Transport in Drosophila-Motoneuronen. Dis Modell Mech. (2015) 8:1389–400. doi: 10.1242/dmm.021246
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
18. Shi GX, Yang WS, Jin L, Materie ML, Ramos JW. RSK2 treibt die Zellmotilität durch Serinphosphorylierung von LARG und Aktivierung von Rho-GTPasen an. In: Proc Natl Acad Sci USA. (2018) 115:E190–9. doi: 10.1073/pnas.1708584115
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar