Frontiers in Pediatrics
Contexte
Un nombre croissant d’études ont démontré que RPS6KA3 est une étiologie moléculaire du syndrome de Coffin–Lowry (CLS) (1, 2), un syndrome semi-dominant lié à l’X qui a été signalé pour la première fois par Coffin en 1966 et caractérisé par une petite taille, un dysmorphisme facial, une déficience intellectuelle grave à profonde (ID), un retard de développement moteur, déformations squelettiques progressives, troubles dentaires, malformations auditives et autres malformations congénitales chez les hommes, avec une intellect allant de normale à sévère altéré chez les femelles hétérozygotes (3-6).
La protéine responsable de RPS6KA3 est le polypeptide 3 de la protéine ribosomique S6 kinase (RSK2), qui est une sérine/thréonine kinase d’une famille de protéines kinases activées par les mitogènes (4). La protéine RSK2 est composée de deux domaines kinases fonctionnels qui sont activés de manière séquentielle par une série de phosphorylations, telles que le site d’amarrage PDK (4). RSK2 s’est avéré être lié à l’incitation à la prolifération cellulaire, au blocage de la différenciation cellulaire et à la protection des cellules contre l’apoptose (7, 8). À ce jour, il y a eu des rapports de cas présentant des symptômes différents et présentant plus de 128 mutations distinctes dans le gène RSK2 divisé en 22 exons de chrXp22.2 (RPS6KA3) (4, 5). Récemment, les mutations de RPS6KA3 se sont avérées liées à une maladie cardiaque, à un ostéosarcome, à une compression du foramen magnum, à des épisodes de chute, etc. (7, 9-12).
Dans le monde, la prévalence spéculative de la CLS peut être de 1/50 000 à 1/100 000 et environ 70 à 80% des probands n’ont pas d’antécédents familiaux, tandis que 20 à 30% ont plus d’un membre de la famille affecté (13). Pour l’instant, il n’existe aucun remède contre cette pathologie. Ici, nous rapportons une mutation de RPS6KA3 à Xp22 chez un garçon chinois. L’aspect phénotypique était typique, y compris le retard de croissance et de développement, comme cela a été décrit dans les rapports précédents.
Présentation du cas
Le proband est un garçon de 12 mois présentant un dysmorphisme facial typique, une déficience auditive et une anomalie osseuse (tableau 1). Il est né après une grossesse normale et a accouché avec un poids de naissance de 2,9 kg (10e percentile) et une longueur de naissance de 45 cm (3e percentile) à 38 semaines, par rapport aux normes de croissance des enfants de l’OMS en 2006. L’aspect facial présente un front bombé, des oreilles proéminentes, des yeux largement espacés, des fissures palpébrales inclinées vers le bas, un nez court avec une large columelle, des alae nasi et un septum épais, un dessous épais et éveillé (Figures 1A, B). Les dents caduques ont éclaté à l’âge de 8 mois (sans retard) (Figure 1C). Les mains sont courtes, charnues et avec des doigts remarquablement hyperextensibles qui s’effilent de larges à étroits avec de petites phalanges terminales et des ongles (Figures 1D, 2D). Mais il n’y avait pas de déformation de son foramen magnum ou de sa colonne vertébrale (Figures 2B, C). Le poids à 12 mois est de 8,2 kg et la taille est de 68.2 cm (< -3,17 z score, OMS). Le métabolisme osseux et l’IGF-1α sont perturbés (Vit D 45,2 nmol / L, IGF-1α < 25 ng / mL). Il a commencé à s’asseoir seul à l’âge de 9 mois et ne pouvait rester sans aide jusqu’à l’âge de 12 mois. À l’âge de 12 mois, son quotient intellectuel (QI) était de 56 selon les calendriers de développement de Gesell. Il avait de la difficulté à rester assis ou à se concentrer pendant la fin de la tâche. Son seuil auditif de réponse auditive du tronc cérébral (ABR) est > 85 db et est diagnostiqué comme un trouble auditif. L’imagerie par résonance magnétique (IRM) a montré la dilatation des ventricules bilatéraux et une diminution de la substance blanche cérébrale (Figure 2A).

Tableau 1. Le phénotype typique du syndrome de Coffin–Lowry de proband.
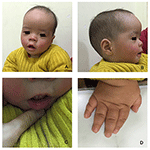
Figure 1. Les dysmorphismes faciaux distinctifs d’un garçon atteint du syndrome de Coffin–Lowry. Une vue latérale du visage du patient (B), des oreilles proéminentes, des yeux largement espacés, des fissures palpébrales inclinées vers le bas, un nez court avec une large columelle, des alae nasi et un septum épais, un dessous épais et éveillé (A, C). (D) Les mains sont courtes et avec un doigt effilé gonflé, de petites phalanges terminales et des ongles.
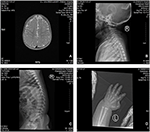
Figure 2. L’imagerie par résonance magnétique (IRM) et les images radiographiques du cas du syndrome de Coffin-Lowry. (A) L’IRM du développement cérébral; (B, C) Photographies radiographiques du canal cervical et du canal rachidien. (D) hyperplasie nodulaire de l’extrémité de la queue du doigt.
Pour l’analyse génétique, des échantillons de sang ont été obtenus de l’individu. La mère avait donné son consentement éclairé pour ses enfants. Cette recherche a été approuvée par le comité de bioéthique pour l’analyse des gènes humains de l’Université du Zhejiang.
L’analyse de séquençage de nouvelle génération (NGS) a été réalisée à l’aide du panel du génome humain Agilent (Agilent Technologies, Inc, Santa Clara, CA, USA). Un c. 2185 C > T à chrX20173554 a été détecté dans le proband, ce qui peut causer p.Mutation Arg729Trp et instabilité RSK2 (Figure 3). Une analyse ultérieure n’a pas détecté la même mutation ponctuelle chez d’autres membres de la famille, y compris la mère et le frère (Matériel supplémentaire). Cela indique que la micromutation est nouvelle mais non héritée de la mère.
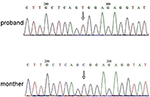
Figure 3. Validation du séquençage de Sanger pour la mutation de RPS6KA3 dans le proband au chromosome Xp22. Une mutation de c. 2185 C > T a été détectée dans la région témoin d’impression de l’allèle 290-300, qui n’a pas été détectée chez la mère de proband.
Discussion
Le cas que nous présentons pour l’évaluation génétique est un garçon de 12 mois né après une grossesse sans incident de parents en bonne santé. Cependant, son âge de développement a été retardé. Comme d’autres patients atteints de CLS, il présentait le phénotype typique observé dans le CLS, notamment une déficience intellectuelle, un développement moteur retardé, une petite taille, des doigts effilés, une déficience auditive et des traits faciaux caractéristiques.
RPS6KA3 (OMIM 303600) est connu pour être muté chez les patients atteints du syndrome de Coffin-Lowry. Exome c.La variante 2185 C > T à Xp22 était nouvelle, séparée de la maladie et était prévue dommageable à l’aide de cinq logiciels in silico différents (SIFT, Polyphen, MutationTaster, FATHMM, LRT).
La hauteur héritée de notre proband est de 167,5 cm car la hauteur de son père est de 173 cm tandis que celle de sa mère est de 150 cm (14). On ne sait toujours pas si la petite taille du syndrome de Coffin–Lowry pourrait être traitée avec des analogues de l’hormone de croissance. Les RSK sont des sérine/thréonine kinases activées par ERK/MAPK et constituent quatre isoformes (RSK1-4) (15). Les RSK agissent comme effecteurs en aval de la signalisation RTK/Ras/ERK (Figure 4) (15-17). En tant que gène responsable de CLS, RSK2 joue un rôle important dans la prolifération et la migration des cellules. L’activation complète de RSK2 nécessite une phosphorylation sur plusieurs sites. Les facteurs de croissance peuvent stimuler la voie du signal ERK vers RSK2 phosphorylé. Comme Ramos l’a rapporté, l’activation d’ERK/ RSK2 est impliquée dans la régulation de la migration des cellules cancéreuses (18).
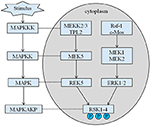
Figure 4. La voie proposée des gènes liés à la RSK. De multiples stimuli pathologiques, tels que la malnutrition intra-utérine, déclenchent des cascades de signalisation en aval de MAPK, qui modulent la prolifération cellulaire et la minéralisation osseuse.
De plus, l’absence de RSK2 est liée à une minéralisation osseuse anormale et induit également des malformations de la colonne vertébrale et des calcifications du ligamentum flavum, telles que la lordose thoracique, la scoliose, la cyphose et la discopathie dégénérative (19, 20). Des rapports antérieurs ont identifié l’ERK/RSK2 comme régulateur de la formation osseuse in vivo (7). En effet, comme Marques et al. des souris déficientes en RSK2 ont récapitulé la perte osseuse progressive due à une diminution de la minéralisation osseuse par les ostéoblastes observée chez les patients atteints de CLS (21). Bien que la différenciation de ces cellules in vitro ait été drastiquement bloquée, le phénotype in vivo a été clairement attribué à une diminution autonome cellulaire de l’activité des ostéoblastes plutôt qu’à une diminution de leur nombre (7). Fait intéressant, il a été récemment démontré que RSK2 interagissait avec le TNF-RI pour affecter la différenciation des ostéoblastes (18, 22). Comme nous le savons, l’hormone de croissance peut entraîner une augmentation de la densité minérale osseuse. Les progrès peuvent aggraver les calcifications du ligamentum flavum et la déformation squelettique (23).
Remarques finales
La présente étude est la première à rapporter un cas chinois de CLS avec mutation de RPS6KA3, ainsi qu’un retard de croissance distinct, de multiples anomalies faciales, des handicaps intellectuels et moteurs. Nous supposons que l’application d’analogues de l’hormone de croissance peut augmenter l’incidence et le caractère invasif des cancers ainsi que le risque de malformation de la colonne vertébrale. Si son efficacité et son innocuité ne sont pas confirmées, l’application d’analogues de l’hormone de croissance sur les patients atteints de CLS doit être envisagée avec une prudence particulière. Des études à long terme avec des résultats pertinents seront essentielles dans la recherche clinique future.
Consentement du patient
Nous avons obtenu un consentement éclairé pour la publication de ce rapport de cas et pour l’utilisation d’images connexes des parents du patient.
Contributions de l’auteur
JS a largement contribué à la conception de ce manuscrit. YL, LZ, JZ et DW ont contribué à l’acquisition, à l’analyse ou à l’interprétation des données. YL a rédigé le manuscrit. JS a révisé de manière critique le manuscrit pour son contenu intellectuel important.
Financement
Ce travail a été soutenu par des subventions de la Fondation Nationale des Sciences naturelles de Chine (n ° 81501293 et 81773440). Nous reconnaissons la famille du cas décrit dans ce rapport qui est au courant et approuve ce compte dépersonnalisé.
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
L’examinateur BP et l’éditeur de gestion ont déclaré leur affiliation commune au moment de l’examen.
Matériel supplémentaire
Figure S1. Validation du séquençage de Sanger pour la mutation de RPS6KA3 chez le frère du proband.
1. Stornetta RL, Zhu JJ. Signalisation Ras et Rap dans la plasticité synaptique et les troubles mentaux. Neuroscientifique (2011) 17:54-78. doi: 10.1177/1073858410365562
Résumé PubMed / Texte intégral CrossRef / Google Scholar
6. Knish LV. Effet des préparations de vitamine E utilisées pendant la grossesse sur le placenta. Pédiatre Akus Ginekol. (1966) 2:51–5.
Résumé PubMed / Google Scholar
10. Gschwind M, Foletti G, Baumer A, Bottani A, Novy J. État épileptique récurrent non convulsif chez un patient atteint du syndrome de coffin-lowry. Syndrome de Mol. (2015) 6:91–5. doi: 10.1159/000430429
Résumé PubMed / Texte intégral CrossRef / Google Scholar
12. Il s’agit d’un produit de qualité et de qualité. Syndrome de Coffin-Lowry et cardiomyopathie ventriculaire gauche sans compaction avec un schéma restrictif. Am J Med Genet A (2011) 155A:3030–4. doi: 10.1002/ ajmg.a. 33856
Résumé PubMed / Texte intégral CrossRef / Google Scholar
14. Cameron N. Programmes de base pour l’évaluation de la maturité squelettique et la prédiction de la taille adulte à l’aide de la méthode Tanner-Whitehouse. Ann Hum Biol. (1984) 11:261–4. doi: 10.1080/03014468400007151
Résumé PubMed / Texte intégral CrossRef / Google Scholar
15. Roméo Y, Zhang X, Roux PP. Régulation et fonction de la famille des protéines kinases RSK. Biochem J. (2012) 441:553-69. doi: 10.1042/BJ20110289
Résumé PubMed / Texte intégral Croisé / Google Scholar
17. Beck K, Ehmann N, Andlauer TF, Ljaschenko D, Strecker K, Fischer M, et al. La perte du gène RSK2 associé au syndrome de Coffin-Lowry modifie l’activité ERK, la fonction synaptique et le transport axonal chez les motoneurones Drosophiles. Modèle Mech. (2015) 8:1389–400. doi: 10.1242/ dmm.021246
Résumé PubMed / Texte intégral CrossRef / Google Scholar
18. Shi GX, Yang WS, Jin L, Matter ML, Ramos JW. RSK2 entraîne la motilité cellulaire par phosphorylation de la sérine de LARG et activation des Rho GTPases. Proc Natl Acad Sci États-Unis. (2018) 115: E190-9. doi: 10.1073/pnas.1708584115
Résumé PubMed / Texte intégral Croisé / Google Scholar