Frontiers in Pediatrics
háttér
egyre több tanulmány bizonyította, hogy az RPS6KA3 a coffin–Lowry szindróma (CLS) (1, 2) molekuláris etiológiája, egy X-kapcsolt félidomináns szindróma, amelyet először Coffin jelentett 1966-ban, alacsony termet, arc diszmorfizmus, súlyos-mély értelmi fogyatékosság (ID), motoros fejlődési késleltetés, progresszív Csontvázdeformitások, fogászati rendellenességek, halláskárosodás és egyéb veleszületett deformitás férfiaknál, az értelem normálistól a súlyosan károsodott heterozigóta nőknél (3-6).
az RPS6KA3-ért felelős fehérje a riboszomális fehérje S6 kináz polipeptid 3 (RSK2), amely a mitogén-aktivált protein-kinázok családjának szerin/treonin kináza (4). Az RSK2 fehérje két funkcionális kináz doménből áll, amelyeket szekvenciálisan aktiválnak egy sor foszforiláció, például PDK dokkolóhely (4). Az RSK2 összefüggésbe hozható a sejtproliferációval, a sejtdifferenciálódás gátlásával és a sejtek apoptózis elleni védelmével (7, 8). A mai napig különböző tünetekkel járó esetekről számoltak be, amelyek több mint 128 különálló mutációt mutattak az RSK2 génben, amelyek 22 exonra osztódtak chrXp22.2 (RPS6KA3) (4, 5). Nemrégiben kiderült, hogy az RPS6KA3 mutációi összefüggenek a szívbetegséggel, az osteosarcomával, a foramen magnum kompresszióval, a csepp epizódokkal stb. (7, 9-12).
világszerte a CLS feltételezett prevalenciája 1/50 000-1/100 000 lehet,A probandok körülbelül 70-80% – ának nincs családtörténete, míg 20-30% – nak egynél több érintett családtagja van, amint arról beszámoltak (13). Ennek a patológiának még nincs gyógymódja. Itt jelentjük az RPS6KA3 mutációját az Xp22-nél egy kínai fiúban. A fenotípusos megjelenés jellemző volt, beleértve a növekedés és a fejlődés késleltetését, amint azt az előző jelentések leírták.
eset bemutatása
a proband egy 12 hónapos fiú, tipikus arc diszmorfizmussal, halláshibával és csontos rendellenességgel (1.táblázat). Normális terhesség után született, születési súlya 2,9 kg (10.percentilis) és születési hossza 45 cm (3. percentilis) a 38. héten, összehasonlítva a WHO 2006-os gyermeknövekedési normáival. Az arc megjelenése domború homlokot, kiemelkedő füleket, széles szemeket, lefelé ferde palpebrális repedéseket, rövid orrot széles columellával, vastag Alae nasi és septum, vastag és elfordított alsó ajkát (1A., B. ábra) mutat. A lombhullató fogak 8 hónapos korban törtek ki (nem késleltetett) (1C ábra). A kezek rövidek, húsosak, és rendkívül hiperextenzív ujjakkal rendelkeznek, amelyek szélestől keskenyig kúposak, kis terminális ujjpercekkel és körmökkel (1D, 2D ábrák). De nem volt deformitás a foramen magnum vagy a gerincoszlop (2b,C ábra). A 12 hónapos súly 8,2 kg, magassága 68.2 cm (<-3,17 Z pontszám, WHO). A csontanyagcserét és az IGF-1-et (vit D) 65,2 nmol/L, IGF-1 6279> 25 ng/mL. 9 hónapos korában kezdett egyedül ülni, és 12 hónapos koráig nem tudott állni segítség nélkül. 12 hónapos korában intelligenciahányadosa (IQ) 56 volt a Gesell fejlődési ütemterve szerint. Nehezen tudott ülni vagy koncentrálni a feladat elvégzése során. Az auditív agytörzsi válasz (ABR) hallási küszöbértéke >85 db, és hallási rendellenességként diagnosztizálják. A mágneses rezonancia képalkotás (MRI) a bilaterális kamrák dilatációját és kevesebb agyi fehérállományt mutatott (2a ábra).

táblázat 1. A proband Coffin–Lowry szindróma tipikus fenotípusa.
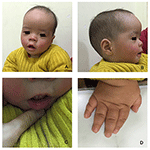
1. ábra. A Coffin–Lowry szindrómás fiú jellegzetes arcdiszmorfizmusai. A beteg arcának oldalnézete (B), kiemelkedő fülek, széles szemkörnyék, lefelé ferde palpebralis repedések, rövid orr széles columellával, vastag Alae nasi és septum,vastag és kifordított alsó ajak (A, C). (D) a kezek rövidek és puffadt, kúpos ujjakkal, kis terminális ujjpercekkel és körmökkel rendelkeznek.
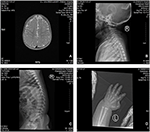
2. ábra. A mágneses rezonancia képalkotás (MRI) és röntgenfelvételek a Coffin-Lowry–szindróma esetéről. (A) az agyi fejlődés MRI-je; (B,C) nyaki és gerinccsatorna röntgenfelvételek. (D) az ujj farok végének noduláris hiperpláziája.
a genetikai elemzéshez vérmintákat kaptunk az egyéntől. Az anya tájékozott beleegyezést adott gyermekeinek. Ezt a kutatást a Zhejiang Egyetem Humán génelemzési bioetikai Bizottsága hagyta jóvá.
következő generációs szekvenálás (NGS) analízist végeztünk Agilent Humán Genom panel (Agilent Technologies, Inc, Santa Clara, CA, USA). A C.2185 C > T a chrX20173554-nél detektálták a probandban, ami p-t okozhat.Arg729Trp mutáció és rsk2 instabilitás (3.ábra). A későbbi elemzés nem mutatta ki ugyanazt a pontmutációt más családtagokban, beleértve az anyát, a testvért (Kiegészítő anyag). Ez azt jelezte, hogy a mikromutáció új, de nem örökölt az anyától.
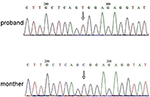
3. ábra. Sanger szekvenálás validálása az RPS6KA3 mutációjára a proband az Xp22 kromoszómán. A C.2185 C > T mutációját a 290-300 allélból származó kontroll régióban detektálták, amelyet proband anyjában nem mutattak ki.
Vita
a genetikai értékeléshez bemutatott eset egy 12 hónapos fiú, aki egészséges szülők eseménytelen terhessége után született. Fejlődési kora azonban késett. A többi érintett CLS-beteghez hasonlóan a CLS-ben megfigyelt tipikus fenotípus volt, beleértve az értelmi fogyatékosságot, a retardált motoros fejlődést, a kis testtartást, a kúpos ujjakat, a halláshibát és a jellegzetes arcvonásokat.
az RPS6KA3 (OMIM 303600) ismert, hogy Coffin-Lowry szindrómában szenvedő betegeknél mutálódik. Exome c.A 2185 C > T változat az Xp22-nél újszerű volt, elkülönült a betegségtől, és az előrejelzések szerint öt különböző In silico szoftver (szitál, Polyphen, MutationTaster, FATHMM, LRT) alkalmazásával káros lehet.
probandunk örökölt magassága 167,5 cm, mivel apja magassága 173 cm, míg anyja magassága 150 cm (14). Még mindig nem világos, hogy a Coffin–Lowry-szindróma alacsony termete kezelhető-e növekedési hormon analógokkal. Az RSK-k az ERK/MAPK által aktivált szerin/treonin kinázok, amelyek négy izoformát alkotnak (RSK1–4) (15). Az RSK-k az RTK/Ras/ERK jelzés downstream effektoraként működnek (4.ábra) (15-17). A CLS – ért felelős génként az RSK2 fontos szerepet játszik a sejtproliferációban és a migrációban. Az RSK2 teljes aktiválása több helyen foszforilezést igényel. A növekedési faktorok stimulálhatják az ERK jelútját a foszforilezett RSK2 felé. Amint Ramos beszámolt róla, az ERK/RSK2 aktiválása részt vesz a rákos sejtek migrációjának szabályozásában (18).
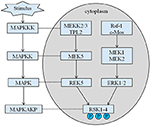
4. ábra. Az RSK-hez kapcsolódó gének javasolt útja. Több kóros inger, mint például az intrauterin alultápláltság, kiváltja a MAPK downstream jelátviteli kaszkádjait, amelyek modulálják a sejtproliferációt és a csont mineralizációját.
mi több, RSK2 ki a funkció kapcsolódik csont mineralizáció abnormális és indukál gerinc fejlődési rendellenességek és meszesedés ligamentum flavum, mint például a mellkasi lordosis, gerincferdülés, kyphosis, és degeneratív betegség lemez (19, 20). Korábbi jelentések az ERK/ RSK2-t a csontképződés szabályozójaként azonosították in vivo (7). Valóban, mint Marques et al. beszámoltak arról, hogy az RSK2-hiányos egerek összefoglalták a progresszív csontvesztést a csökkent csont mineralizáció miatt a CLS-ben szenvedő betegeknél megfigyelt osteoblastok (21). Bár ezeknek a sejteknek az in vitro differenciálódása drasztikusan blokkolódott, az in vivo fenotípus egyértelműen az oszteoblasztok aktivitásának sejt autonóm csökkenésének tulajdonítható, nem pedig számuk csökkenésének (7). Érdekes módon az RSK2 nemrégiben kimutatták, hogy kölcsönhatásba lép a TNF-RI-vel az érintett oszteoblasztok differenciálódásához (18, 22). Mint tudjuk, a növekedési hormon fokozott csont ásványi sűrűséget okozhat. Az előrehaladás súlyosbíthatja a ligamentum flavum meszesedését és a csontváz deformitását (23).
Záró megjegyzések
ez a tanulmány az első, amely az Rps6ka3 mutációval rendelkező CLS Kínai esetét, valamint egyértelműen növekedési retardációt, többszörös arc rendellenességeket, értelmi és motoros fogyatékosságot jelentett. Úgy gondoljuk, hogy a növekedési hormon analógok alkalmazása növelheti a rákos megbetegedések előfordulását és invazivitását, valamint a gerinc malformáció kockázatát. Ha hatásosságát és biztonságosságát nem erősítik meg, a növekedési hormon analógok CLS-ben szenvedő betegeknél történő alkalmazását különösen óvatosan kell mérlegelni. A releváns kimenetelű hosszú távú vizsgálatok elengedhetetlenek lesznek a jövőbeni klinikai kutatásban.
beteg beleegyezése
tájékozott beleegyezést kaptunk az esetjelentés közzétételéhez és a beteg szüleitől származó kapcsolódó képek felhasználásához.
szerzői hozzájárulások
js jelentősen hozzájárult a kézirat koncepciójához. Az YL, LZ, JZ és DW hozzájárult az adatok megszerzéséhez, elemzéséhez vagy értelmezéséhez. YL elkészítette a kéziratot. JS kritikusan felülvizsgálta a kéziratot a fontos szellemi tartalom érdekében.
finanszírozás
ezt a munkát a kínai Nemzeti Természettudományi Alapítvány támogatásai támogatták (81501293 és 81773440). Elismerjük a jelentésben leírt eset családját, amely ismeri és jóváhagyja ezt az azonosítatlan fiókot.
összeférhetetlenségi nyilatkozat
a szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.
a BP recenzens és a handling Editor a felülvizsgálat idején közös hovatartozásukról nyilatkoztak.
Kiegészítő Anyag
S1 Ábra. Sanger szekvenálás validálása az RPS6KA3 mutációjára a proband testvérében.
1. Stornetta RL, Zhu JJ. Ras és Rap jelátvitel szinaptikus plaszticitásban és mentális zavarokban. Idegtudós (2011) 17: 54-78. doi: 10.1177/1073858410365562
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
6. Knish LV. a terhesség alatt alkalmazott E-vitamin készítmények hatása a placentára. Pediatr Akus Ginekol. (1966) 2:51–5.
PubMed Absztrakt / Google Scholar
10. Gschwind M, Foletti G, Baumer a, Bottani a, Novy J. visszatérő nonconvulzív status epilepticus coffin-lowry szindrómás betegben. Mol Syndromol. (2015) 6:91–5. doi: 10.1159/000430429
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
12. Martinez HR, Niu MC, Sutton VR, Pignatelli R, Vatta M, Jefferies JL. Coffin-Lowry szindróma és a bal kamrai noncompaction cardiomyopathia korlátozó mintázattal. Am J Med Genet A (2011) 155A:3030–4. doi: 10.1002 / ajmg.a. 33856
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
14. Cameron N. alapvető programok a csontváz érettségének értékelésére és a felnőtt magasságának előrejelzésére Tanner-Whitehouse módszerrel. Ann Hum Biol. (1984) 11:261–4. doi: 10.1080/03014468400007151
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
15. Romeo Y, Zhang X, Roux PP. Az RSK protein kinázok családjának szabályozása és működése. Biochem J. (2012) 441:553-69. doi: 10.1042 / BJ20110289
PubMed absztrakt / CrossRef teljes szöveg / Google Tudós
17. Beck K, Ehmann N, Andlauer TF, Ljaschenko D, Strecker K, Fischer M, et al. A Coffin-Lowry szindrómához kapcsolódó rsk2 gén elvesztése megváltoztatja az ERK aktivitását, szinaptikus funkcióját és axonális transzportját a Drosophila motoneuronokban. Dis Modell Mech. (2015) 8:1389–400. doi: 10.1242 / dmm.021246
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós
18. Shi GX, Yang WS, Jin L, anyag ML, Ramos JW. Az RSK2 a LARG szerin foszforilezésével és a Rho Gtpázok aktiválásával hajtja végre a sejtek motilitását. Proc Natl Acad Sci USA. (2018) 115:E190–9. doi: 10.1073/pnas.1708584115
PubMed Absztrakt / CrossRef Teljes Szöveg / Google Tudós