Frontiers in Pediatrics
Tło
rosnąca liczba badań wykazała, że RPS6KA3 jest etiologią molekularną zespołu Coffina–Lowry ’ ego (CLS) (1, 2), semidominacyjnego zespołu związanego z X, który został po raz pierwszy zgłoszony przez Coffina w 1966 r. i charakteryzuje się niskim wzrostem, dysmorfizmem twarzy, ciężką do głębokiej niepełnosprawnością intelektualną (ID), opóźnienie, postępujące deformacje szkieletowe, zaburzenia uzębienia, wada słuchu i inne wrodzone deformacje u mężczyzn, o wartości od normalnej do ciężkiej zaburzenia u heterozygotycznych samic (3-6).
białkiem odpowiedzialnym za RPS6KA3 jest polipeptyd 3 kinazy rybosomalnej S6 (rsk2), która jest kinazą serynowo-treoninową z rodziny kinaz białkowych aktywowanych mitogenem (4). Białko RSK2 składa się z dwóch funkcjonalnych domen kinazowych, które są aktywowane sekwencyjnie przez szereg fosforylacji, takich jak miejsce dokowania PDK (4). Stwierdzono, że RSK2 jest związany z proliferacją komórek, blokowaniem różnicowania komórek i ochroną komórek przed apoptozą (7, 8). Do tej pory istnieją doniesienia o przypadkach z różnymi objawami i z ponad 128 odrębnymi mutacjami w genie RSK2 podzielonym na 22 eksony chrXp22.2 (RPS6KA3) (4, 5). Ostatnio stwierdzono, że mutacje RPS6KA3 są związane z chorobami serca, kostniakomięsakiem, ściskaniem otworu magnum, epizodami kropli i tak dalej (7, 9-12).
na całym świecie spekulowana częstość występowania CLS może wynosić 1/50 000-1/100 000 i około 70-80% badanych nie ma historii rodzinnej, podczas gdy 20-30% ma więcej niż jednego dotkniętego członka rodziny, jak podano (13). Jak dotąd nie ma lekarstwa na tę patologię. Tutaj zgłaszamy mutację RPS6KA3 w Xp22 u chińskiego chłopca. Wygląd fenotypowy był typowy, w tym opóźnienie wzrostu i rozwoju, jak opisano w poprzednich raportach.
Prezentacja przypadku
proband jest 12-miesięcznym chłopcem z typowym dysmorfizmem twarzy, wadą słuchu i wadami kości (Tabela 1). Urodził się po prawidłowej ciąży i poród miał masę urodzeniową 2,9 kg (10 percentyl) i długość urodzeniową 45 cm (3 percentyl) w 38 tygodniu, w porównaniu ze standardami wzrostu dziecka WHO w 2006 roku. Wygląd twarzy przedstawia wybrzuszone czoło, wydatne uszy, szeroko rozstawione oczy, skośne szczeliny powiekowe, krótki nos z szeroką kolumellą, grube alae nasi i przegrodą,grube i everted underlip (ryc. 1A, B). Zęby Liściaste wybuchły w wieku 8 miesięcy (bez opóźnień) (ryc. 1C). Dłonie są krótkie, mięsiste i mają wyjątkowo rozciągliwe palce, które zwężają się od szerokich do wąskich z małymi końcowymi paliczkami i paznokciami (rys. 1D, 2D). Ale nie było deformacji otworu magnum ani kolumny kręgosłupa(ryc. 2b, C). Waga w wieku 12 miesięcy wynosi 8,2 kg, a wzrost 68.2 cm (< -3,17 z, WHO). Metabolizm kości i IGF-1α jest zaburzeniem (Vit D 45,2 nmol/L, IGF-1α < 25 ng/mL). Zaczął siedzieć sam w wieku 9 miesięcy i nie mógł stać bez pomocy aż do 12 miesiąca życia. W wieku 12 miesięcy jego iloraz inteligencji (IQ) wynosił 56 według schematu rozwoju Gesella. Miał trudności z pozostaniem w pozycji siedzącej lub koncentracji podczas wykonywania zadania. Jego próg słuchowy odpowiedzi pnia mózgu (ABR) wynosi >85 db i jest diagnozowany jako zaburzenie słuchu. Obrazowanie metodą rezonansu magnetycznego (MRI) wykazało rozszerzenie obustronnych komór i zmniejszenie ilości materii Białej w mózgu (ryc. 2A).

Tabela 1. Typowy fenotyp zespołu Coffina–Lowry ’ ego probanda.
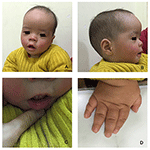
Rysunek 1. Charakterystyczne dysmorfizmy twarzy chłopca z zespołem Coffin-Lowry. Widok z boku twarzy pacjenta (B), wydatne uszy, szeroko rozstawione oczy, w dół skośne szczeliny powiekowe, krótki nos z szeroką kolumellą, grube alae nasi i przegroda,gruby i everted podlip (A, C). (D) ręce są krótkie i z podpuchniętym stożkowym palcem, małymi końcowymi paliczkami i paznokciami.
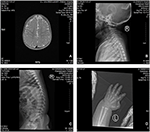
Rysunek 2. Rezonans magnetyczny (MRI) i zdjęcia rentgenowskie przypadku zespołu Coffina-Lowry ’ ego. A) MRI rozwoju mózgu; B, C) zdjęcia rentgenowskie kanału szyjki macicy i rdzenia kręgowego. (D) guzkowy rozrost końca ogona palca.
do analizy genetycznej pobrano próbki krwi od osobnika. Matka udzieliła świadomej zgody swoim dzieciom. Badania te zostały zatwierdzone przez Komitet bioetyki do analizy genów ludzkich na Uniwersytecie Zhejiang.
analizę sekwencjonowania nowej generacji (NGS) przeprowadzono przy użyciu panelu genomu ludzkiego Agilent (Agilent Technologies, Inc, Santa Clara, CA, USA). W probandzie wykryto c.2185 C > T w chrX20173554, co może powodować P.Mutacja Arg729Trp i niestabilność RSK2 (ryc. 3). Późniejsza analiza nie wykazała tej samej mutacji punktowej u innych członków rodziny, w tym matki, brata (materiał uzupełniający). Wskazywało to, że mikromutacja jest nowa, ale nie odziedziczona po matce.
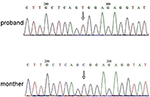
Rysunek 3. Walidacja sekwencjonowania Sangera dla mutacji RPS6KA3 w probandzie na chromosomie Xp22. Mutacja c.2185 C > t została wykryta w regionie kontrolnym imprintingu z allelu 290-300, czego nie wykryto u matki probanda.
dyskusja
przypadek, który prezentujemy do oceny genetycznej, to 12-miesięczny chłopiec, który urodził się po niezakłóconej ciąży od zdrowych rodziców. Jego wiek rozwojowy był jednak opóźniony. Podobnie jak inni chorzy na CLS, miał typowy fenotyp obserwowany w CLS, w tym niepełnosprawność intelektualną, opóźniony rozwój ruchowy, mały wzrost, zwężające się palce, wada słuchu i charakterystyczne rysy twarzy.
RPS6KA3 (OMIM 303600) jest znany z mutacji u pacjentów z zespołem Coffina-Lowry ’ ego. Exome C.Wariant 2185 C > T w Xp22 był nowatorski, oddzielony od choroby i przewidywano, że będzie szkodliwy przy użyciu pięciu różnych programów in silico (SIFT, Polyphen, MutationTaster, FATHMM, LRT).
odziedziczony wzrost naszego probanda wynosi 167,5 cm ze względu na wzrost ojca wynosi 173 cm, natomiast wzrostu matki 150 cm (14). Czy niski wzrost zespołu Coffina-Lowry ’ ego można leczyć analogami hormonu wzrostu nadal nie jest jasne. RSKs są kinazami serynowo-treoninowymi aktywowanymi przez ERK / MAPK i stanowią cztery izoformy (RSK1–4) (15). RSKs działają jako efektory sygnalizacji RTK/Ras/ERK (Rysunek 4) (15-17). Jako gen odpowiedzialny za CLS, RSK2 odgrywają ważną rolę w proliferacji i migracji komórek. Pełna aktywacja RSK2 wymaga fosforylacji w wielu miejscach. Czynniki wzrostu mogą stymulować szlak sygnałowy ERK do fosforylowanego RSK2. Jak donosił Ramos, aktywacja ERK / RSK2 bierze udział w regulacji migracji komórek nowotworowych (18).
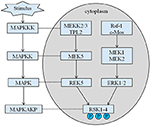
Rysunek 4. Proponowany szlak genów związanych z RSK. Liczne patologiczne bodźce, takie jak niedożywienie wewnątrzmaciczne, wywołują kolejne kaskady sygnałowe MAPK, które modulują proliferację komórek i mineralizację kości.
co więcej, Brak funkcji RSK2 jest związany z nieprawidłową mineralizacją kości, a także indukuje wady rozwojowe kręgosłupa i zwapnienia więzadła flavum, takie jak lordoza klatki piersiowej, skolioza, kifoza i choroba zwyrodnieniowa dysku (19, 20). Wcześniejsze doniesienia wskazywały ERK / RSK2 jako regulator tworzenia kości in vivo (7). Rzeczywiście, jak Marques et al. u myszy z niedoborem RSK2 obserwowano postępującą utratę kości spowodowaną zmniejszoną mineralizacją Kości przez osteoblasty obserwowane u pacjentów z CLS (21). Chociaż różnicowanie się tych komórek in vitro zostało drastycznie zablokowane, fenotyp in vivo wyraźnie przypisywano raczej zmniejszeniu autonomicznej aktywności osteoblastów niż zmniejszeniu ich liczby (7). Co ciekawe, niedawno wykazano, że RSK2 oddziałuje z TNF-RI w celu zróżnicowania osteoblastów (18, 22). Jak wiemy, hormon wzrostu może powodować zwiększoną gęstość mineralną kości. Postęp może nasilać zwapnienia więzadła flavum i deformację szkieletu (23).
Uwagi końcowe
niniejsze badanie jest pierwszym, który zgłosił Chiński przypadek CLS z mutacją RPS6KA3, a także wyraźne opóźnienie wzrostu, wielokrotne nieprawidłowości twarzy, niepełnosprawność intelektualną i motoryczną. Spekulujemy, że stosowanie analogów hormonu wzrostu może zwiększyć częstość i inwazyjność nowotworów, a także ryzyko deformacji kręgosłupa. Jeśli jego skuteczność i bezpieczeństwo nie są potwierdzone, stosowanie analogów hormonu wzrostu u pacjentów z CLS należy szczególnie ostrożnie rozważyć. Długoterminowe badania z odpowiednimi wynikami będą niezbędne w przyszłych badaniach klinicznych.
zgoda pacjenta
uzyskaliśmy świadomą zgodę na opublikowanie tego raportu przypadku i wykorzystanie powiązanych zdjęć od rodziców pacjenta.
wkład autora
js znacząco przyczynił się do powstania tego rękopisu. YL, LZ, JZ i DW przyczyniły się do pozyskania, analizy lub interpretacji danych. YL sporządził rękopis. Js krytycznie zrewidował rękopis pod kątem ważnych treści intelektualnych.
finansowanie
praca ta została wsparta grantami z National Natural Science Foundation Of China (nr 81501293 i 81773440). Przyjmujemy do wiadomości rodzinę przypadków opisanych w niniejszym raporcie, która zna i zatwierdza to de-zidentyfikowane konto.
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
recenzent i redaktor prowadzący zadeklarowali wspólną przynależność w momencie recenzji.
Materiał Uzupełniający
Rysunek S1. Sanger sekwencjonowanie Walidacja dla mutacji RPS6KA3 w proband brat.
1. Stornetta RL, Zhu JJ. Sygnalizacja Ras i Rap w plastyczności synaptycznej i zaburzeniach psychicznych. [2011-01-17 17: 54] doi: 10.1177/1073858410365562
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
6. Knish LV. wpływ preparatów witaminy E stosowanych w ciąży na łożysko. Pediatr Akus Ginekol. (1966) 2:51–5.
PubMed Abstract / Google Scholar
10. Gschwind m, Foletti G, Baumer a, Bottani a, Novy J. nawracający stan bezdrgawkowy u pacjenta z zespołem Coffina-lowry ’ ego. Zespół Molowy. (2015) 6:91–5. doi: 10.1159/000430429
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
12. Martinez HR, NIU MC, Sutton VR, Pignatelli R, Vatta M, Jefferies JL. Zespół Coffina-Lowry ’ ego i kardiomiopatia bez kompakcji lewej komory z restrykcyjnym wzorem. Am J Med Genet a (2011) 155A:3030–4. doi: 10.1002 / ajmg.a. 33856
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
14. Cameron N. BASIC programs for the assessment of skeletal maturity and the prediction of adult height using the Tanner-Whitehouse method. Ann Hum Biol. (1984) 11:261–4. doi: 10.1080/03014468400007151
PubMed Abstract / CrossRef Pełny tekst / Google Scholar
15. Romeo Y, Zhang X, Roux PP. Regulacja i funkcja rodziny kinaz białkowych RSK. Biochem J. (2012) 441: 553-69. doi: 10.1042 / BJ20110289
PubMed Streszczenie / CrossRef Pełny tekst / Google Scholar
17. Beck K, Ehmann N, Andlauer TF, Ljaschenko d, Strecker K, Fischer m, et al. Utrata genu rsk2 związanego z zespołem Coffina-Lowry ’ ego zmienia aktywność ERK, funkcję synaptyczną i transport aksonalny w Motoneuronach Drosophila. Dis Model Mech. (2015) 8:1389–400. doi: 10.1242 / dmm.021246
PubMed Streszczenie / CrossRef Pełny Tekst / Google Scholar
18. Shi GX, Yang ws, Jin L, Matter ML, Ramos JW. RSK2 napędza ruchliwość komórek przez fosforylację SERYNOWĄ LARG i aktywację Rho GTPases. Proc Natl Acad sci USA. (2018) 115:E190–9. doi: 10.1073 / pnas.1708584115
PubMed Abstract / CrossRef Full Text / Google Scholar