Fronteiras em Pediatria
Fundo
Um número crescente de estudos tem demonstrado que RPS6KA3 é uma etiologia molecular do Caixão–Lowry síndrome (CLS) (1, 2), X-linked semidominant síndrome que foi relatada pela primeira vez pelo Caixão, em 1966, e caracterizada por baixa estatura, facial dysmorphism, grave a profunda deficiência intelectual (DI), atraso no desenvolvimento motor, progressivas deformidades esqueléticas, doenças dentárias, audiência defeito e outras deformidades congênitas no sexo masculino, e com a inteligência varia de normal a ser gravemente doentes heterozigóticos (3-6).
A proteína responsável pela RPS6KA3 é a proteína ribossomal S6 quinase polipeptídeo 3 (RSK2), que é uma serina/treonina quinase de uma família de agente mitogénico ativado cinases (4). A proteína RSK2 é composta por dois domínios cinase funcionais que são ativados de forma sequencial por uma série de fosforilações, como o local de acoplagem PDK (4). Verificou-se que o RSK2 está relacionado com o desencadeamento da proliferação celular, o bloqueio da diferenciação celular e a protecção das células da apoptose (7, 8). Até à data, têm sido notificados casos com sintomas diferentes e com mais de 128 mutações distintas no gene RSK2 divididas em 22 exons de chrXp22.2 (RPS6KA3) (4, 5). Recentemente, verificou-se que as mutações do RPS6KA3 estão relacionadas com doença cardíaca, osteosarcoma, compressão do forame magno, episódios de queda e assim por diante (7, 9-12).
em todo o mundo, a prevalência especulada de CLS pode ser de 1/50, 000-1 / 100.000 e cerca de 70-80% dos probands não têm história familiar, enquanto 20-30% têm mais de um membro da família afetado, como relatado (13). Até agora, não há cura para esta patologia. Aqui, reportamos uma mutação do RPS6KA3 no Xp22 num rapaz Chinês. A aparência fenotípica foi típica, incluindo atraso de crescimento e desenvolvimento, como foi descrito nos relatórios anteriores.
Apresentação Do Caso
o proband é um rapaz de 12 meses de idade com dismorfismo facial típico, deficiência auditiva e anormalidade óssea (Tabela 1). Ele nasceu após uma gravidez normal e teve um peso de nascimento de 2,9 kg (10º percentil) e comprimento de nascimento de 45 cm (3º percentil) a 38 semanas, em comparação com os padrões de crescimento infantil da OMS em 2006. A aparência facial apresenta testa saliente, orelhas proeminentes, olhos muito espaçados, fissuras palpebrais inclinadas, nariz curto com columela larga, alae nasi espessa e septo,espesso e com um lábio inferior evertido (figuras 1A, B). Os dentes decíduosforam erupções aos 8 meses de idade (não retardados) (figura 1C). As mãos são curtas, carnudas, e com dedos notavelmente hiperextensíveis que se afunilam de largura a estreita com pequenas falanges terminais e pregos (figuras 1D, 2D). Mas não houve deformidade do forame magnum ou coluna vertebral (figuras 2B,C). O peso aos 12 meses é de 8,2 kg e a altura é de 68.2 cm (<-3, 17 Z score, WHO). O metabolismo ósseo e o IGF-1α são perturbações (Vit D 45, 2 nmol/L, IGF-1α < 25 ng/mL). Ele começou a sentar-se sozinho aos 9 meses e não podia ficar sem ajuda até 12 meses de idade. Aos 12 meses de idade, seu quociente de inteligência (QI) era de 56 De acordo com os esquemas de desenvolvimento Gesell. Ele teve dificuldade em permanecer sentado ou se concentrar durante a conclusão da tarefa. Seu limiar auditivo de resposta ao tronco cerebral auditivo (ABR) é >85 db e é diagnosticado como uma doença auditiva. A ressonância magnética (MRI) mostrou a dilatação dos ventrículos bilaterais e menos matéria branca cerebral (figura 2A).

Quadro 1. O fenótipo típico da síndrome de Coffin-Lowry de proband.
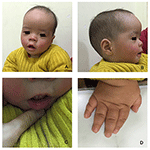
Figura 1. Os distintos dismorfismos faciais de um rapaz da síndrome de Lowry. Uma visão lateral da face do paciente (B), orelhas proeminentes, olhos muito espaçados, fissuras palpebrais inclinadas, nariz curto com colunela larga, alae nasi espessa e septo,espesso e com um lábio inferior evertido (A, C). (D) as mãos são curtas e com o dedo inchado afunilado, pequenas falanges terminais e unhas.
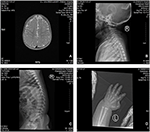
Figura 2. A ressonância magnética (MRI) e imagens de raios-X do caso da síndrome de Coffin–Lowry. A) a ressonância magnética do desenvolvimento cerebral; b,c) fotografias de raios-X do canal Cervical e da coluna vertebral. D) hiperplasia Nodular da extremidade da cauda do dedo.
para análise genética, amostras de sangue foram obtidas do indivíduo. A mãe tinha dado consentimento informado para seus filhos. Esta pesquisa foi aprovada pelo Comitê de Bioética para análise de genes humanos na Universidade Zhejiang.
Next Generation Sequencing (NGS) analysis was performed using Agilent Human Genome panel (Agilent Technologies, Inc, Santa Clara, CA, USA). Um c. 2185 C > T no chrX20173554 foi detectado no proband, o que pode causar p.Mutação Arg729Trp e instabilidade RSK2 (Figura 3). A análise subsequente não detectou a mesma mutação pontual em outros membros da família, incluindo mãe, irmão (material suplementar). Isto indica que a micromutação é nova, mas não herdada da mãe.
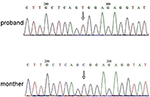
Figura 3. Validação da sequência de Sanger para a mutação do RPS6KA3 no proband no cromossoma Xp22. Foi detectada uma mutação de C. 2185 C > T na região de controlo de impressão do alelo 290-300, que não foi detectada na mãe de proband.
Discussão
O caso que apresentamos para avaliação genética é um menino de 12 meses, que nasceu depois de uma gravidez sem intercorrências do saudável pais. No entanto, sua idade de desenvolvimento foi atrasada. Como outros pacientes afetados pela CLS, ele teve o fenótipo típico observado na CLS, incluindo deficiência intelectual, desenvolvimento motor retardado, pequena estatura, dedos afiados, deficiência auditiva e características faciais.Sabe-se que
RPS6KA3 (OMIM 303600) sofre mutação em doentes com síndrome de Coffin-Lowry. Exome C.2185 c > t variante em Xp22 foi nova, segregada da doença, e foi previsto para ser prejudicial usando cinco diferentes software slico (SIFT, Polyphen, MutationTaster, FATHMM, LRT).
a altura herdada do nosso probando é de 167,5 cm por causa da altura de seu pai é de 173 cm, enquanto a altura de sua mãe é de 150 cm (14). Ainda não está claro se a curta estatura da síndrome de Coffin–Lowry poderia ser tratada com análogos da hormona do crescimento. As RKS são serinas / trionina cinases activadas por ERK / MAPK e constituem quatro isoformas (RSK1–4) (15). Os RSK actuam como operadores a jusante da sinalização RTK/Ras/ERK (Figura 4) (15-17). Como gene responsável pelo CLS, o RSK2 desempenha um papel importante na proliferação celular e migração. A ativação completa do RSK2 requer fosforilação em vários locais. Os factores de crescimento podem estimular a via do sinal ERK para o RSK2 fosforilado. Como Ramos relatou, a ativação de ERK/RSK2 está envolvida na regulação da migração de células cancerosas (18).
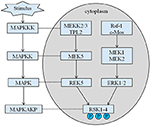
Figura 4. A via proposta dos genes relacionados com a RSK. Múltiplos estímulos patológicos, como desnutrição intra-uterina, desencadeiam cascatas sinalizadoras de MAPK, que modulam a proliferação celular e mineralização óssea.
o Que é mais, RSK2 de função está relacionada com a mineralização óssea anormal e também induz malformações da coluna vertebral e calcificações de ligamentum flavum, tais como torácica, lordose, escoliose, cifose, e doença degenerativa do disco (19, 20). Relatórios anteriores identificaram o ERK / RSK2 como um regulador da formação óssea in vivo (7). De facto, como Marques et al. notificados, ratinhos com deficiência em RSK2 recapitularam a perda óssea progressiva devido à diminuição da mineralização óssea pelos osteoblastos observados em doentes com CLS (21). Embora a diferenciação destas células in vitro tenha sido drasticamente bloqueada, o fenótipo in vivo foi claramente atribuído a uma diminuição da actividade Autónoma celular dos osteoblastos, em vez de uma diminuição do seu número (7). Curiosamente, o RSK2 demonstrou recentemente interagir com o TNF-RI na diferenciação dos osteoblastos afectados (18, 22). Como sabemos, a hormona do crescimento pode causar um aumento da densidade mineral óssea. O progresso pode agravar as calcificações de ligamento flavum e deformidade esquelética (23).O presente estudo é o primeiro a relatar um caso chinês de CLS com mutação de RPS6KA3, bem como retardo de crescimento distintamente, anomalias faciais múltiplas, deficiências intelectuais e motoras. Especula-se que a aplicação de análogos da hormona do crescimento pode aumentar a incidência e a inviabilidade de cancros, bem como o risco de malformação da coluna vertebral. Se a sua eficácia e segurança não forem confirmadas, a aplicação de análogos da hormona do crescimento em doentes com CLS deve ser especialmente cuidadosamente considerada. Os estudos a longo prazo com resultados relevantes serão essenciais na futura investigação clínica.
consentimento do doente
obtivemos consentimento informado para a publicação deste relatório de caso e para a utilização de imagens relacionadas dos pais do doente.
contribuições dos autores
JS contribuíram substancialmente para a concepção deste manuscrito. YL, LZ, JZ e DW contribuíram para a aquisição, análise ou interpretação de dados. YL redigiu o manuscrito. JS revisou criticamente o manuscrito para conteúdo intelectual importante.
financiamento
este trabalho foi apoiado por subvenções da Fundação Nacional de Ciências Naturais da China (n. º 81501293 e 81773440). Reconhecemos a família do caso descrito neste relatório que tem conhecimento e aprova esta conta desidentificada.
Declaração de conflito de interesses
os autores declaram que a investigação foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.
the reviewer BP and handling Editor declared their shared affiliation at the time of review.
Material Suplementar
Figura S1. Validação da sequência de Sanger para a mutação do RPS6KA3 no irmão do proband.
1. Stornetta RL, Zhu JJ. Ras e Rap sinalizando plasticidade sináptica e distúrbios mentais. Neuroscientist (2011) 17:54-78. doi: 10.1177/1073858410365562
PubMed Abstract | CrossRef texto completo | Google Scholar
6. Knish LV. efeito das preparações de vitamina E utilizadas na gravidez na placenta. Pediatr Akus Ginekol. (1966) 2:51–5.
PubMed Abstract / Google Scholar
10. Gschwind M, Foletti G, Baumer a,Bottani a, Novy J. Mol Syndromol. (2015) 6:91–5. doi: 10.1159/000430429
PubMed Abstract | CrossRef texto completo | Google Scholar
12. Martinez HR, Niu MC, Sutton VR, Pignatelli R, Vatta M, Jefferies JL. Síndrome de Coffin-Lowry e cardiomiopatia de não-Acção ventricular esquerda com um padrão restritivo. Am J Med Genet a (2011) 155A:3030–4. doi: 10.1002 / ajmg.A. 33856
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Cameron N. BASIC programs for the assessment of skeletal maturity and the prediction of adult height using the Tanner-Whitehouse method. Ann Hum Biol. (1984) 11:261–4. doi: 10.1080/03014468400007151
PubMed Abstract | CrossRef texto completo | Google Scholar
15. Romeo Y, Zhang X, Roux PP. Regulação e função da família de proteínas cinases RSK. Biochem J. (2012) 441:553-69. doi: 10.1042 / BJ20110289
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Beck K, Ehmann N, Andlauer TF, Ljuschenko D, Strecker K, Fischer M, et al. A perda do gene rsk2 associado à síndrome de Coffin-Lowry altera a actividade ERK, a função sináptica e o transporte axonal em motoneuros de Drosophila. Dis Modelo Mech. (2015) 8:1389–400. doi: 10.1242/dmm.021246
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Shi GX, Yang WS, Jin L, Matter ML, Ramos JW. RSK2 impulsiona a motilidade celular por fosforilação de serina de LARG e ativação de Rho GTPases. Proc Natl Acad Sci USA. (2018) 115:E190–9. doi: 10.1073 / pnas.1708584115
PubMed Abstract | CrossRef Full Text | Google Scholar